|
A
peer-reviewed electronic journal published by the Institute for Ethics and ISSN
1541-0099 26(1)
– January 2016 |

Human Connectome Mapping and Monitoring Using
Neuronanorobots
Nuno R. B. Martins
Center for Mathematics
University of Minho
n-martins@n-martins.com
Wolfram Erlhagen
Center for Mathematics
University of Minho
wolfram.erlhagen@math.uminho.pt
Robert A. Freitas, Jr.
Institute
for Molecular Manufacturing
Journal of Evolution and Technology - Vol. 26 Issue 1 – January 2016 - pgs 1-24
Abstract
Neuronanorobotics
is the application of medical nanorobots to the human brain. This paper
proposes three specific classes of neuronanorobots, named endoneurobots,
gliabots and synaptobots, which together can non-destructively map and monitor
the structural changes occurring on the 86 x 109 neurons and the
2.42 x 1014 synapses in the human brain, while also recording the
synaptic-processed 4.31 x 1015 spikes/sec carrying electrical
functional information processed in the neuronal and synaptic network.
1. Introduction
Preserving
human brain information is essential for truly preserving human life. A crucial
part of that precious information is structural in nature and is referred to as
the human connectome, believed to be the processing framework for
functional information. No currently available technological tool claims the
capacity to preserve whole human brain structural and functional connectome
information comprehensively with proper temporal and spatial resolution.
Any
nonlethal kind of brain damage caused by neurological disorders can
irreversibly damage the human brain, causing loss of precious information. Such
loss may include loss of brain structural information, such as neural
connectivity, or loss of brain functional information, such as neurotransmitter
activity patterns. Loss of information might affect proper cellular and
organ-level neurological functions, eventually altering higher mental states,
personality, and ultimately individuality and self-awareness. The usual causes
of information loss are physical trauma and a variety of degenerative and viral
disorders. Once destroyed and not previously preserved, such information cannot,
even in principle, be recovered using current techniques, thus permanently
diminishing patient health.
Medical
technological tools and methods for monitoring, measuring, validating, and
archiving comprehensive brain-related structural information are urgently
necessary because current technologies do not provide the necessary temporal
and spatial resolution requirements for preserving fundamental human brain
information. For example, none of the current technologies, either destructive
or non-destructive in nature, can monitor the whole human brain in vivo,
in real-time, with adequate cellular and subcellular temporal and spatial
resolution for mapping the brain structural and functional electrical
connectome with appropriate detail.
Medical
nanorobotics technology, a medical technology first examined technically in the
Nanomedicine book series (Freitas 1999, 2003), is expected to permit
measuring, mapping, and monitoring structural and functional human brain
information along with many other medical applications (Freitas 1998, 2000,
2003, 2005a, 2005b, 2005c, 2006a, 2006b, 2007, 2010; Morris 2001; Astier et al.
2005; Patel et al. 2006; Park et al. 2007; Popov et al. 2007; Martel et al.
2009; Mallouk and Sen 2009; Kostarelos 2010; Mavroides and Ferreira 2011).
Neuronanorobotics, involving a specific class of medical nanorobots, is
expected to permit in vivo, whole-brain, real-time mapping and
monitoring of the relevant structural and functional cellular and sub-cellular
brain information. Such nanorobots should, for example, permit mapping the
whole human brain connectome (Sporns et al. 2005; Lu et al. 2009; Anderson et
al. 2011; Kleinfeld et al. 2011; Seung 2011) by the coordinated activities of
three primary types of cooperating neuronanorobots proposed here, to be called
endoneurobots, gliabots, and synaptobots, as described below.
2.
Structural and functional connectome-associated information
To
comprehensively restore a brain to a proper healthy structural and functional
state will likely require more than a simple connectomics neuro-synaptic
diagram, as current neuroscience literature points to several other relevant
forms of structural brain information. Consequently, medical technology might
have to be capable of preserving information at the cellular and subcellular
level, performing the following tasks:
• detecting and characterizing each of the individual 86.06 ± 8.12 billion neurons (Azevedo et al. 2009; Herculano-Houzel 2009) with volumes from ~33.5 micron3 (for granule cell) up to ~4.18 x 106 micron3 (for motor neurons);
• monitoring the loss of ~100,000 neurons a day (~1 per second) (Pakkenberg and Gundersen 1997; Arking 2006);
• monitoring five phases of neuron genesis and elimination processes, occurring on time scales between 1-10 days (Gross 2000);
• monitoring the activity of the 84.61 ± 9.83 x 109 glial cells (Azevedo et al. 2009);
• monitoring axon initial segment (typically the first 50 μm of axon) and axon hillock molecular characterization, particularly the quantification of Kv1 family potassium channel types responsible for shaping action potential waveforms, with turnover of ~3 hours (Lu et al. 2004; Palmer and Stuart 2006; Kole et al. 2007);
• monitoring the 10,000-52,000 micron long dendritic trees each having surface areas of 700-54,000 µm2 (Donohue and Ascoli 2008);
• monitoring of dendritic development, establishment, and remodeling, and monitoring important neuron organelles, especially the neuron nucleus (diameter 4-100 µm); and
• monitoring intracellular compartments.
Comprehensive
structural connectome monitoring might also require technology capable of
detecting synaptic plasticity structural changes induced by synaptic
cross-talk, such as synaptic based long-term potentiation, long-term
depression, short-term plasticity, metaplasticity and homeostatic plasticity,
as well as measuring the dynamic composition of the post-synaptic density, each
having ~10,000 proteins comprising ~100 different types (Sheng and Hoogenraad
2007).
It
is not clear if human brain functional information can be deduced solely from
the underlying structural information, so functional information remains either
essential or important in providing redundant measurement and inference for
brain information preservation. Such functional information may include in
vivo measurement of action potential associated electrical information;
neurotransmitters, hormones, and neurotrophic-based extracellular communication
(e.g., synaptic, paracrine, or endocrine signaling); intracellular chemical
communication (e.g., via GTP-binding proteins, second messenger molecules,
protein kinases, ion channels, and other effector proteins); and molecular
diffusion occurring in the 20 per cent of brain volume comprising the
extracellular spaces (Thorne and Nicholson 2006; Sandberg and Bostrom 2008).
Intracellularly,
it might be relevant to identify and quantitatively measure:
• neuron gene expression and transcription factors by monitoring the 20,000-25,000 protein-coding genes and 2.85 billion nucleotides (IHGSC 2004), with total information size of ~3.08 gigabytes (IHGSC 2004);
• neuron nucleus traffic of the 22,000-505,000 molecules of mRNA per neuron cell (Islam 2011);
• local protein synthesis and degradation in dendrites, axons and synapses;
• action-potential-induced openings of the ~20 Ca2+ channels present, on average, per each active zone, measuring consequent fast release with a delay 50-500 μsec (Sabatini and Regehr 1996; Südhof 2004);
• mechanisms of calcium wave propagation at velocity ~28 μm/sec including diffusion of IP3 and extracellular ATP (velocity of ~41 μm/sec) (Newman 2001), among other functional and structural information, along with the resultant Ca2+ transient (lasting 400-500 μsec, with a>5 μM Ca2+) (Südhof 2004);
• the 17-500 nm diameter synaptic vesicles (average ~40 nm) (Qu et al. 2009; Südhof 2004), with the average lumen volume of 11,500 nm3 in CNS (“typical” synaptic terminals have in the order of ~100 vesicles per active zone (Smith et al. 2008) with the vesicular content extruded in 0.1-100 ms (Travis and Wightman 1998));
• synaptic vesicle molecular content (between 5,000 and 106 molecules) (Kandel et al. 1991; Südhof 2004), enumerating the different vesicle pools along with the respective depletion times and refill mechanisms (Rollenhagen and Lübke 2006);
• synaptic vesicle proteic and lipidic composition, typically 40% phosphatidylcholine, 32% phosphatidylethanolamine, 12% phosphatidylserine, 5% phosphatidylinositol, and 10% cholesterol (Südhof 2004);
• the “typical” 20 nm ± 8 nm synaptic cleft of chemical synapses (Qu et al. 2009);
• fast (~250-400 mm/day), intermediate (~15-50 mm/day), and slow (~0.1-4 mm/day) axonal transport (Perrot et al. 2008); and
• axon growth and axon elimination.
On
the borderline between functional and structural monitoring, a controversial
question is the importance of monitoring various chemical concentration changes
in minimal-size volumetric compartments of, say, 125 nm3 (5 nm x 5
nm x 5 nm). At this scale, changes are at the most fundamental size limit for
computational brain simulation. Neuronanorobots will not monitor every single
brain volumetric compartment, but might monitor select volumetric compartments
with functional relevance. It remains to be determined how much of this
potentially large quantity of structural and functional connectome information
is actually essential to enable the computational reconstruction of the human
connectome.
3.
Recent results in human connectome mapping
The human “connectome” is
defined as the full set of human brain neurons and their network of synaptic connections.
There is not yet a consensus that the connectome holds the structural
correlates of personality and individuality, but it is clear that the human
connectome holds valuable human information (Seung 2010). The essential neuronal and synaptic subcomponent
building blocks to reconstruct the whole-brain structural and
functional connectome remains unknown, but a list of the cellular and
molecular components used in computational neural
microcircuit connectome reconstruction can be seen elsewhere (Markram,
2006)
Mapping a human
whole-brain connectome with cellular and synaptic resolution is the ultimate
goal of some current research efforts, and several milestones have already been
achieved on the path to solve this grand challenge, as summarized below.
Several
small nonhuman brain connectomes have already been preserved computationally,
in part or in whole, using destructive and non-destructive techniques.
The
first to be mapped, and the best-known publicly, is the C. elegans
connectome. This effort produced a comprehensive synapse-level nervous system
reconstruction using a series of 5000 serial thin (70-90 nm) sections,
including the scanning of the 170 neuron processes and the 64 muscles (White et
al. 1986; Chen et al. 2006; Varshney 2011; Jarrell 2012). The online platform “Worm Atlas” contains
details on all C. elegans systems, including its connectome data (White
et al. 1986; Wormatlas 2008; Varshney et al. 2011).
Also
scanned and “uploaded” was
the connectome of the predatory nematode Pristionchus pacificus. The Pristionchus
pacificus connectome was compared with the C. elegans connectome
(Bumbarger 2013) and some serious similarities appeared. For example, both the
pharyngeal nervous systems of the Pristionchus pacificus and the C.
elegans are composed of a network of 20 identified neurons of 14 cell
types. The numeric similarities were associated with differences on the
synaptic connectivity mapping of the pharyngeal nervous systems of two
nematodes. The differences were proven to be correlated with divergent feeding
behavior (Izquierdo and Beer 2013), which can be considered as the first “proof” that brain
connectomics information is at least a part of the fundamental structural brain
information required for preserving high-level behaviors.
The
mouse primary visual cortical connectome (spanning layers 1, 2/3, and upper
layer 4) has already been computationally reconstructed, employing a procedure
that included structural connectome scanning, using electron microscopy based
technology, and functional connectome scanning, using two-photon microscopy to
determine the functional properties
of about 14 of the cells of that same volume of tissue. This provided a voxel
resolution of45 µm x
4 µm x
4 µm
for a total tissue volume of approximately 450 µm x
350
µm x 50 µm (Bock
et al. 2011).
A structural/functional
data project built a neocortical microcircuit database (Ramaswami 2015) with
neuron data that included 135 electrophysiological parameters (derived from
responses to stimuli), 234 geometric parameters (based on neural reconstruction),
and 51 genetic properties (from single cell RT-PCR data). For synapses the data
included some physiological properties and 15 parameters for anatomical
properties, 5 parameters for synaptic dynamics, and 14 parameters for kinetics.
The
inner plexiform layer of the mammalian retina connectome was mapped with ~2 nm
resolution using automated transmission electron microscope imaging, generating
a 16.5 terabyte connectome data set. The data represents a column of tissue
0.25 mm in diameter, spanning the inner nuclear, inner plexiform, and ganglion
cell layers of the rabbit (Anderson et al. 2011). Data sets from mouse cortex
with a 3 nm x 3 nm x 30 nm spatial resolution had previously yielded 660
gigabytes of images (Kasthuri et al. 2009).
Diffusion
imaging (DI) technology, applied on living animals and humans, has permitted
mapping long-range projections of small clusters of neurons (without synaptic
resolution) (Mori and Zhang 2006; Kasthuri and Lichtman 2007; Bohland et al.
2009; Clayden 2013; Craddock et al. 2013). High-resolution
diffusion tensor imaging of the human optic chiasm was performed ex vivo at
156 μm resolution
in-plane, but the typical DI resolution is on the millimeter range. Whole-brain
mesoscale connectomes (i.e. “projectomes”) that are scanned in vivo by DI technologies
do not contain structural information about individual neuronal cells, and also
don’t permit following single axonic processes or
identifying individual synapses.
A
spinal cord brain map project is close to completion, and the Paul Allen
Institute just finished the first phase of a human brain atlas, a four-year
project started in 2010 (Allen 2009). A proposed five-year, $20 million
mesoscopic scale mouse connectivity brain map is also underway (Bohland 2009;
Modha-blog 2009).
Two-photon
calcium imaging combined with large-scale electron microscopy of serial thin
sections permits physiological and anatomical reconstruction of a group of
neurons in the mouse primary visual cortex (Bock 2011).
Whole
brain gene expression information at cellular level – the gene expression map – was computationally reconstructed from histology, with
pixel size 0.95 μm2,
and from MRI data, voxel size 12.3 μm3 (Lein et al. 2007; Jones 2011; Allen Institute 2014;
Allen Institute-mouse 2014).
CLARITY
enables estimations of the joint morphological statistics of most neurons in a
tissue sample at the same time (Chung 2013; Chung and Deisseroth 2013).
In a
project focused on functional data monitoring, detailed system level maps of a
human individual’s “functional connectome” were compiled using resting-state functional MRI
(R-fMRI) that gathered data from 1,414 brains (Biswal et al. 2010; FCP 2010).
As
for achievements at the computational level, daydreaming was simulated by a
computational model (Deco et al. 2013). Still at the computational level, IBM
claims to have simulated 530 billion neurons and 100 trillion synapses on the
world’s fastest computer (Theodore et al. 2012).
That
is ~5 times the number of neurons of a human brain and is very near the total
number of synapses of a human brain (est. 2.42 x 1014) (Martins et
al. 2012). It is not clear what they define as a synapse or what the
capabilities of those synapses are, but these networks might soon start
demonstrating human level subsystem data processing output in limited domains
(e.g., visual recognition) using massive parallelization. The computational
timescale is ~1500 times slower than the human brain timescale (i.e., 1 second
of brain time currently takes ~25 minutes to simulate) but this doesn’t seem to be a problem because most real brain
processing/decision happens in seconds (Theodore et al. 2012).
One
open source project has the goal of preserving the human connectome with
synaptic level resolution (Connectome Project 2011).
The
human whole-brain “generic” connectome project has huge implications for
Artificial Intelligence and the well-known “Singularity” (e.g., Kurzweil 2005). Two billion-dollar projects are
already underway to solve this challenge: the “Human Connectome Project” and
the “Brain Research Through Advancing Innovative
Neurotechnologies initiative” (BRAIN) (NIH News
2009; Human Brain Project 2013).
The
U.S. government called the 1990s the “Decade
of the Brain”
(Jones and Mendell 1999), but the first
serious “call to action” for
a coordinated effort to collect, archive, and disseminate the crucial
information of the human brain connection matrix was made in 2005 (Sporns et
al. 2005; Ascoli 2009). Eight years later, U.S. national agencies launched a
formal call for mapping the whole human brain activity map as part of the
challenge for functional connectomics (Alivisatos et al. 2012; Kandel et al.
2013).
Destructive
structural 3D computational reconstruction of a whole human brain with 20 µm resolution (average cellular level resolution) has
already been done, nearly preserving the first complete whole human brain
neuron soma cytoarchitectural anatomy (Amunts et al. 2013). The next goal was
stated to be the creation of a brain model with a spatial voxel resolution of
approximately 1 µm to capture details
of single cell morphology. This is intended to integrate gene expression data
from the Allen Institute human brain project. Other projects are taking the
effort of mapping the whole human brain connectome with synaptic connections
included, and the remaining challenges seem surpassable in the decades ahead
(Connectome Project 2011).
Brain
tissue structural scanning tools generate tens of petabytes of data for mapping the 1 cm3 mouse brain with
nanometric resolution. As no fully automated tools are currently available, the
posterior 3D brain tissue reconstructions currently have to be manually handled
(Helmstaedter et al. 2008). In the last 40 years the improvements on the speed
of brain scanning and image reconstruction have grown exponentially, and if
this trend persists a whole human brain reconstruction should happen in the
decades ahead (Kurzweil 2005).
4.
Non-destructive vs. destructive scanning techniques
Non-destructive structural whole brain monitoring techniques in the
form of computerized scanning-based imaging modalities, such as positron
emission tomography (PET) and magnetic resonance imaging (MRI), provide
non-destructive three-dimensional views of the brain with ~1 mm resolution,
with clinical MRI scan voxel resolution typically sized 1 mm x 1 mm x 3 mm
(Johnson 2010; Kandel et al. 2000). Such resolution permits regional analyses
of brain structure but is clearly insufficient for investigation of structures
underlying intercellular communication at the level of individual neurons or
synapses (Fiala and Harris 2001). Still at the whole brain level, a
non-destructive technique called high-definition fiber tractography provides
accurate reconstruction of white matter fiber tracts, but again with a clearly
insufficient resolution of ~1 mm (Shin et al. 2012).
Micro-CT
scanners, with a typical scan time between 10 minutes and 2 hours, provide
high-resolution tomography of specimens up to a few centimeters in diameter,
with the highest spatial resolution being 2 µm,
not enough to detect most synapses. The state-of-the-art tomographic nano-CT
scanners (the micro-CT successor) achieves structural resolutions between
500-50 nm. Currently, there are only three main nano-CT scanners available
commercially: the Nanotom, the SkyScan-2011, and the Xradia nanoXCT. The
Nanotom provides a resolution of ~500 nanometers pixels, and handles maximum
object size of 150 mm height and 120 mm diameter (GE-MCS 2013) (roughly the
size of a whole human brain). The SkyScan-2011 has a slightly better resolution
of ~400 nm pixels and similar object size constraints. The Xradia nanoXCT
claims to be capable of providing a spatial resolution between 300-50 nm
(Tkachuk et al. 2007). Although the nano-CT scanner resolutions might permit
extraction of some cellular detail and eventually identification of some
synapses, a lot of structural information remains un-captured and, most
problematically, the technology does not permit in vivo brain scanning.
Destructive scanning technologies, by contrast, can achieve
near-nanometric structural scanning resolution permitting visualization of
individual synapses and their constituents. In a type of destructive brain
scanning, an automated ultramicrotome produces large amounts of nanometric
30-100 nm thick sections, which are then scanned by either a transmission
electron microscope (TEM) or a serial block-face scanning electron microscopy
(SBEM), or by an optical microscope (Hayworth et al. 2006; Anderson and Itallie
2009; Kasthuri et al. 2009; Hayworth 2012). After scanning, posterior software
reconstruction uses specialized software, such as RESCOP or KNOSSOS, to trace
neuronal connections (Helmstaedter et al. 2011) via individual neuron tagging
using fluorescent proteins (aka. brainbow technique), which facilitates the
analysis of neuronal circuitry (and mapping of glial territories) on a large
scale, and helps on structural cellular level and neuronal circuitry
information analysis (Livet et al. 2007; Lichtman et al. 2008). A different
high-throughput technique called BOINC (also known as “barcoding of individual neuronal connections”) permits establishing circuit connectivity at single
neuron and synaptic resolution using high-throughput DNA sequencing (Zador et
al. 2012).
For
small size samples, and to avoid the technical difficulties and the laborious
process involved in ultrastructural electron microscopy based techniques, X-ray
nanotomography with precision down to 30 nm permits avoiding chemically
fixation, staining or cutting cells (Helmholtz Association 2010). It can
deliver high-resolution 3-D images of the entire fast-frozen cell, but it still
takes weeks to be applied in one cell (Helmholtz Association 2010). For single
cells, scanning light confocal microscopy provides three-dimensional views of
individual neurons down to about 1 μm of resolution.
The
current destructive approaches provide near-nanometric atomic structural
resolution and seem to be a promising tool for mapping the “generic” whole
human brain connectome, provided the speed of scanning a certain brain volume
maintains historical improvement rates over the next decades. Although current
scanned volumes are far from a whole human brain volume, a process to achieve a
whole human brain is envisionable (Hayworth 2012).
There
are at least four inherent problems with existing destructive scanning
approaches.
First,
it is not clear if the different biomolecular machinery can be distinguished by
the next generation of these techniques.
Second,
although functional information might not be necessary (e.g., if atomic
structural resolution were achieved), functional information is not captured by
these techniques.
Third,
the structural connectome forms the basis
for the processing of most functional information that flows in the brain. The
interplay between structural and functional information forms the brain as a
whole, and although likely, it is unknown if comprehensive structural
whole-brain information is sufficient to preserve human life and individuality.
Brain simulations are expected to help elucidate the coupling between
structural connectivity and functional data, culminating a crucial step to
understand the human brain (Friston 2011; Bargmann and Marder 2013).
Fourth,
destructive techniques are expected to face resistance from patients during
implementation in clinical practice, because the maintenance of consciousness
continuity is uncertain or unknown.
In
summary, destructive scan technologies are nearer than non-destructive methods
to the technical goal of preserving whole human brain connectome information,
likely providing enough structural resolution to create at least a static map
of a now-disassembled brain. But there are major limitations and serious
uncertainties surrounding its applicability in future medical practice with
live human patients. Also, while destructive “bottom up” approaches permit
nanometric spatial resolution, they cannot easily be applied to whole human
brains. For example, electron microscopy permits reconstruction with nanometric
resolution, but acquisition rates are too low for the mapping of large volumes
(Briggman and Bock 2012).
On
the other hand, current non-destructive or “top-down” approaches can scan the whole brain but lack the
required spatial resolution. For example,
diffusion imaging can image large fiber bundles in vivo across the whole
brain but the resolution is too coarse to detect single neuronal processes,
with DI non-specificity limiting the classification of fiber bundles (Clayden
2013).
The
proposed new neuronanorobotics approach appears uniquely to offer whole-brain
nanometric scan resolution on a non-destructive basis.
5.
Neuronanorobot mission and conceptual design
Neuronanorobotic
functional and structural connectome monitoring includes two main tasks: (1)
monitoring the functional action potential based electrical information
processing happening in synapses and neurons; and (2) monitoring the synaptic
and neuronal structural changes associated with the processing of such
electrolytic-based functional data. Two
conceptually different approaches can be used to complete these two tasks
successfully, with the main difference
between strategies residing on the navigation strategy (intracellular
navigation or extracellular navigation) and nanorobot positioning strategy
(intracellular monitoring or extracellular monitoring). In the proposed mission
design, structural and functional connectome monitoring can be done
intracellularly using three types of neuronanorobots and an installed in
vivo fiber optic network (described elsewhere: Freitas 1999). The three
types of neuronanorobots – called
endoneurobots, gliabots, and synaptobots – are to be stationed, respectively, in the neuron soma, in the glial
cells, and near a pre- or post-synaptic side of a synapse (or a set of
synapses).
5.1.
Rationale for intracellular navigation and monitoring
Multiple
considerations appear to favor the choice of intracellular, rather than
extracellular, navigation and monitoring by the three different types of
neuronanorobots:
(1)
Extracellular space is extremely tight, with the average width of
the free brain extracellular space (ECS) lying between 38-64 nm
(Thorne and Nicholson 2006). ECS width around the cell bodies of
oligodendrocytes typically measures 0.3-10 nm; the ECS around the
cell bodies of astrocytes oscillates from 4-154 nm. By comparison, a
synaptobot is 500 nm in its smallest dimension.
(2) The
ECS is thoroughly infiltrated by the extracellular matrix, a structure composed
of negatively charged glycosaminoglycans and proteoglycans (Syková 2008) which play a major role in orchestrating the
development of the CNS, are involved in remodeling the
adult CNS after injury, and are involved in events that may underlie some
aspects of memory (Syková 2008). These extracellular space proteins, if seriously disrupted,
might cause a cascade of intracellular signaling, triggering neuron epigenetic
and structural changes. For example, the application of tension forces to
cultured neurons or vascular smooth muscle cells via ECM adhesions results in
force-dependent increases in microtubule polymerization (Ingber 2006; Lelièvre 2009).
(3) Each
synaptobot might serve between 1-10 synapses, implying a fleet of 24-242 x 1012
robots to cover the 2.42 x1014
synapses in the human brain (Martins et al. 2012) corresponding to a total
fleet volume of 1.2-12 x 1013 µm3
or 12-120 cm3, representing ~0.9-9% of total human brain volume.
This lies within the 1-10% “safe” tissue and organ intrusiveness limit recommended
elsewhere (Freitas 2003). If parked
extracellularly, this volume of synaptobots would correspond to 4-50% of the
extracellular space, likely interfering with, for example, the dynamics of
extracellular based-communication, potentially causing considerable neuron
epigenetic changes, and potentially causing force-dependent increases in
microtubule polymerization. Synaptobots parked intracellularly occupy a much
safer fraction of 1.25%-12.5% of the brain intracellular space.
(4)
The neuron intracellular environment offers numerous biological
proof-of-principle examples of circulating cellular bionanomachinery. Many of
these –
such as resident mitochondria, synaptic
vesicles, and other interior organelles – have similar dimensions to the circulating synaptobots, navigate
similar routes, and park in similar regions. Aside from microglia (see below),
the extra-cellular environment is generally reserved for chemical circulation.
(5)
Apart from relatively slow neural axon growth, there is no good biological
example of large organelles or cells frequently moving around the brain ECS.
Extracellular microglia rarely circulate, are mobilized only after injury,
infection, or disease, and are otherwise by default in a resting state. Microglial movements do not represent a massive
volume moving in the extracellular space, because microglia constitute only
~20% of the total 85 x 109 glial population in the CNS (Kreutzberg
1995; Azevedo et al. 2009) and are distributed with no significant local
differences in the white and grey matter of the brain (Kreutzberg 1995).
(6)
Extracellular navigation and monitoring by synaptobots would require a complex
communication protocol for establishing a correspondence between neurons and
the monitored synapses. With intracellular navigation and monitoring, synapses
are by default associated with the appropriate neuron.
We
conclude that intracellular navigation and monitoring seem much more
appropriate for neuronanorobots. The extracellular space should be used only
when intracellular navigation is not possible. This may occur in a few special
instances where a small percentage of synaptobots might have to temporarily
navigate through the extracellular space to avoid extremely thin axon diameters
(Section 5.6).
5.2
Endoneurobots and gliabots
Endoneurobots
and gliabots are the larger of the proposed neuronanorobots, each with 10 µm3 volume. In their mission, endoneurobots
leave the bloodstream to enter the brain parenchyma, and navigate inside the
neuropil until they enter the neuron cell soma and get positioned
intracellularly in the axon initial segment (AIS). Similarly, gliabots leave
the bloodstream, enter the respective glial cell, and position themselves
intracellularly in the most appropriate intra-glial region, which varies.
Since
action potentials might be initiated in different cellular sub-compartments,
endoneurobots will be parked at the AIS (the most likely spot for action
potential initiation) where they will monitor the large majority of action
potentials. In neurons where some action potentials are initiated at the first
nodes of Ranvier or the axon hillock, two synaptobots placed at the first node
of Ranvier and at the axon hillock can ensure proper action potential waveform
detection.
Endoneurobots
monitor action potential based electrical information using FET-based
nanosensors existent on the endoneurobot surface (Martins et al. 2015). For
monitoring neuronal structural changes (some caused by the processing of action
potentials) and once securely anchored to the neuron internal membrane surface
(“typical” neurons
have a volume of 14,000 µm3 or (~24
µm)3), endoneurobots might employ a tactile
scanning probe to image the surrounding membrane surface area of (1.4 micron)2
in ~2 sec to ~1 nm2 resolution (~1 mm/sec tip velocity), or ~50 sec
to ~0.2 nm (e.g., atomic) resolution (~0.2 mm/sec tip velocity) assuming a scan
rate of ~106 pixels/sec (Freitas 1999).This same scanning tool will
be used by all three types of robots.
Endoneurobots
and gliabots also provide communication (and data transmission) support for
synaptobots.
5.3
Synaptobots
Synaptobots
are clearly the most technically challenging of the three neuronanorobots, due
to: (1) the small volume of the robots, (2) the navigational requirements
necessary to get to the monitoring position, and (3) the challenge of scanning
data from synaptic structures. Communication (and data transmission) support is
provided by endoneurobots, freeing synaptobots from the necessity of including
this high-volume support machinery within their onboard structure.
The
task of monitoring raw data traffic for an entire living human brain minimally
requires a network data handling capacity of (5.52 ± 1.13) x 1016 bits/sec, corresponding to an
estimated synaptic-processed spike rate of (4.31 ± 0.86) x 1015 spikes/sec (Martins 2012).
This is the event detection requirement for the entire embedded in vivo
sensor system in the context of nanorobotic whole-brain monitoring.
(Non-electrical channels must also be recorded but the data processing
requirements for these additional channels are far less demanding: Martins
2012). An in vivo nanorobotic auxiliary fiber optic system with 30 cm3
volume and generating 4-6 watts waste heat (Freitas 1999; Freitas 2010) is
capable of handling 1018 bits/sec, which enables rapid data transfer
(including the action potential waveform) and real-time brain-state monitoring
(Freitas 2010; Martins 2012). This auxiliary fiber optic system, coupled with
the support for data transmission by endoneurobots and gliabots, minimizes the
demand for onboard information storage in synaptobots. The synaptobot
nanocomputer might be composed of a 0.01 µm3
CPU device capable of >100 megaflops, with 10 onboard copies for redundancy.
This is comparable to other nanorobot designs with similar degrees of mission
complexity, such as the microbivore with its 0.11 µm3 onboard computer and memory storage
system (Freitas 2005c).
Synaptobots
are the smallest of the three nanorobot types, having a volume of 0.5 µm3. Synaptobots are responsible for
monitoring synapses, the most challenging and important sub-cellular structures
in the human brain. Synapses (either the 5-25% electrical or the 75-95% chemical
variety (DeFilipe and Fariñas 1992)) are part of
the neural network that processes human brain information. They play a crucial
role in brain information processing (IBM 2008) and are involved in learning
and memory (Black et al. 1990; Bliss and Collingridge 1993; Holtmaat and Svoboda 2009; Liu 2012), long-term and
short-term memory storage and deletion (Kandel 2001; Lee et al. 2008), and
temporal information processing (Fuhrmann et al. 2002), and are the key
elements for signal transduction and plasticity in the human brain (Rollenhagen
and Lübke 2006; Rollenhagen et al. 2007). Synapses are so
important that proper synapse formation during childhood provides the substrate
for cognition, whereas improper formation or function leads to neuro-developmental
disorders, including mental retardation and autism (McAllister 2007). The loss
of synapses, as occurs in Alzheimer’s
patients, is highly related to cognitive decline (Dekosky and Scheff 1990;
Terry et al. 1991; Scheff and Price 2006).
In
the exemplar synaptobot mission, the robots enter the human body via the
bloodstream, later cross the blood-brain barrier to enter the brain parenchyma,
and finally enter the respective neuron soma. This process is mediated by the
support of auxiliary transport nanorobots.
The
synaptobots are delivered via brain vasculature to avoid long-distance
navigation inside the brain parenchyma. Auxiliary transport nanorobots of
volume ~20 µm3 (~3.2 x 2.5 x 2.5 µm) each convey cargos of 24 synaptobots (~12 µm3) through the circulatory system and into
the neuron soma. The full complement of synaptobots are transported by a fleet
of ~1 trillion auxiliary transport nanorobots, which perform ~10 round trips to
complete the insertion of all synaptobots in the first medical intervention.
Each neuron, on average, receives ~117 cargos for an average total delivery of
(2.42 x 1014 synapses / 86 x 109 neurons ≈) 2800 synaptobots, assigning one robot per synapse
(Martins et al. 2012). The protocol for regularly updating the number of synaptobots
in the brain (due to robot damage, synapse elimination, neuron death, new
synaptic formation, etc.) is started by the endoneurobots which communicate the
necessities. The ~1 trillion auxiliary transport nanorobots should suffice to
handle the workload of dynamically adjusting the physical deployments of
synaptobots.
The
protocol that auxiliary transport nanorobots may use to cross the BBB and
traverse the neuropil will be similar to the protocol employed by the
endoneurobots and the gliabots, due to the similar diameters of auxiliary
transport nanorobots (2.5 µm) and the
endoneurobots and gliabots (2.2 µm).
Afterwards,
the synaptobots either stay in the neuron soma or navigate from the neuron soma
along the axon or dendrite into a pre- or post-synaptic structure. This is the
place where synaptic monitoring will occur. To identify the pre- and
post-synaptic structures of synapses, synaptobots must necessarily map, from
inside the cell, the surfaces of the neuron axon (for axo-axonic, axo-somatic, and
axo-dendritic synapses), the soma (for somato-axonic, somato-somatic, or
somato-dendritic synapses), and the dendrites (for dendro-somatic,
dendro-axonic, and dendro-dendritic synapses). During this process, synaptobots
will locate a large variety of synapses in the brain, with the most abundant
synapses in the central nervous system being the asymmetric (excitatory)
axo-spinous synapses (Harris 1999).
Once
at the monitoring position near a pre- or post-synaptic structure, the
functional component of the synaptobot mission is to record the exact timing
and intensity of the action potential electrical information arriving at the
synapses and to monitor the associated changes happening on key structural
elements of the synapse. With one synaptobot positioned at each of the 2.42 x
1014 human brain synapses (Martins et al. 2012), action potential
data is acquired using ~3375 nm3 FET-based neuroelectric nanosensors
(Martins et al. 2015) permitting monitoring of the synaptic-processed 4.31 x 1015
spikes/sec. Data collection will have a temporal resolution of at least 0.1 ms,
which is enough for waveform characterization even at the highest human neuron
firing rates of 800 Hz. Assisted by the other two nanorobot types
(endoneurobots and gliabots), the synaptobots then transmit the 5.52 x 1016
bits/sec of continuous action potential data (Martins et al. 2012) into the
associated in vivo fiber optic networking system.
As
for the structural component of the synaptobot mission, mapping and subsequent
monitoring of relevant neuron structure will be done using tactile scanning
probe nanosensors (Freitas 1999) with special scanning tips that permit
synaptic bouton volume and shape to be measured, along with other relevant synaptic
structural characteristics. An ideal structural scanning process would also
permit monitoring the main ultrastructural components of a chemical synapse,
whether located in the presynaptic axon terminal, the synaptic cleft, or the
post-synaptic terminal. Of primary relevance is the structural information
encoded in postsynaptic density (PSD). Other possibly relevant sources of
structural information are the active zone (AZ), synaptic vesicles (coated
vesicles, dense core vesicles, and double-walled vesicles), endoplasmic
reticulum, mitochondria, and punctum adhaerens (PA).
Ideally
the tactile nanosensors will permit measurement of:
(1)
the volume, shape, and organelle content of the synaptic boutons.
(2)
the postsynaptic density (PSD) at each synapse, allowing the robot to identify
major protein components of the different brain PSDs (comprising up to ~1461
different proteins) (Bayés 2010) with each PSD
having on average a total of ~10,000 proteins, typically ~100 copies of ~100
different proteins (Sheng and Hoogenraad 2007).
(3)
the functional information flowing through the “typical”
20 nm ± 8
nm synaptic cleft of chemical synapses (Qu 2009).
By
monitoring synaptic structural changes, the neuronanorobots will also be
monitoring synaptic plasticity and cross-talk, including synaptic based
long-term potentiation, long-term depression, short-term plasticity,
metaplasticity, and homeostatic plasticity.
5.4
Mitochondrial proof of principle
Synaptobots
will have an independent locomotion system for moving along axons and dendrites
in both directions. While mitochondria depend on an existing neuronal transport
system, mitochondria distribution and mobility patterns provide a reasonable
biological proof-of-principle of biocompatibility for synaptobots moving along
the neuronal processes. Mitochondria and synaptobots are similar in length and
volume, with axonal mitochondria existing as discrete organelles typically 1-3 μm in length (Trushina et al. 2012; Sheng 2014), 0.75 µm in diameter, and 0.5 µm3 in volume (Miyamoto 1986), very close to robot
dimensions. The anticipated robot deployment linear number density of ~0.5
synaptobots/µm-length of axonic or dendritic process is
similar to the typical 0.2-0.4 mitochondria/µm-length
in NTG mouse axons (Miyamoto 1986). The deployment volumetric number density of
0.5 synaptobots/µm3 of
axonic or dendritic process seems acceptable because the more distal dendrites
of CA1 hippocampal neurons and the ultrastructural features of synaptic
mitochondria indicate a numeric density of 0.8 mitochondria/µm3 for mitochondria in the most distal
dendrites and in principle the most inaccessible areas of dendrites (Bertoni-Freddari
et al. 2006).
The
activity levels required for synaptobots are similar to the mobility behaviors
exhibited by mitochondria. Synapses have high demand for local mitochondrial
ATP production, so the distribution of mitochondria to all neuronal regions
requires a bidirectional axonal and dendritic transport system that can deliver
and retain these organelles to the appropriate axonal and dendritic regions
with high energy requirements (Schwarz 2013). The transport system involves
kinesin and dynein motors along with an exceptional number of signaling
pathways and regulatory proteins that interact with the dendritic or axonal
cytoskeleton. Mitochondria are moved along axons and dendrites bidirectionally
over long distances, frequently changing direction, and being stabilized by the
actin network. In mature neurons, 10-40% of axonal mitochondria are in motion
at any one time – more
specifically, 54.07% ± 2.53% of axonal
mitochondria measured in syntaphilin knockout mice are stationary and sitting
out of synapses, 16.29 ± 1.66% are docking at
synapses, 14.77 ± 1.58% are motile and
passing through synapses, 7.01 ± 1.29% are pausing at
synapses for short periods of time (<200 s), and 8.30 ± 1.52% are pausing at synapses for long period of time
(>200 s) (Sun et al. 2013).
Synaptobot
maximum velocities of ~1 µm/s are also expected
to respect biocompatibility requirements. Bidirectional movements of
mitochondria in axons and dendrites are reported at velocities ranging from
0.32-0.91 µm/s (Morris and Hollenbeck 1995; MacAskill et al.
2009), and mitochondrial motility in NTG neurons is reported as 0.93 ± 0.55 µm/sec for anterograde
motion and 0.97 ± 0.63 µm/sec for retrograde motion (Trushina et al. 2012).
5.5
Nanorobot volumetric intrusiveness in the brain
The “average” human
intracranial volume is estimated as 1700 cm3 (Rengachary 2005), being ~1350 cm3 of brain
cells (Freitas 1999) (typically with “average” dimensions of ~140 x 167 x 93 mm), ~150 cm3
of blood, and ~150 cm3 of CSF (Rengachary
and Ellenbogen 2005). Experimental data provides a considerably wider range for
measured intracranial volumes (1152-1839 cm3) and for total average
CSF (82-125.3 cm3), with total brain cell parenchyma volume of 1319
cm3 including 489 cm3 of white matter and 786 cm3
of gray matter (Vaidyanathana et al. 1997; Nopoulos et al. 2000). The typical
human brain is estimated to have (86.06 ± 8.2)
x 109 neurons, with the largest (80.2%) percentage or (69.03 ± 6.65) x 109 neurons located in the
cerebellum, the second largest (19%) percentage or (16.34 ± 2.17) x 109 neurons located in the cerebral
cortex, and the remaining smallest (0.8%) percentage or (0.69 ± 0.12) x 109 neurons in the rest of the
brain (Azevedo et al. 2009).
Inside
each neuron will reside one 10 µm3 endoneurobot,
a total robot volume of 0.86 cm3 representing ~0.06% of total human
brain volume. Parking one 10 µm3 gliabot
in each of the 84.6 ± 9.8 x 109
glial cells in the human brain (Azevedo et al. 2009) adds another ~0.06% of human brain volume. In each case, the lone nanorobot
represents <0.1% of a “typical” ~14,000 µm3
neuron volume, and thus is minimally intrusive.
The
largest robot intrusion on brain volume occurs with insertion of the
synaptobots. How many synaptobots are needed? About 90 per cent of all synapses
are in dendritic spines, with “typical” number
density of spines ranging from 1-10 spines/µm
length of dendrite, depending on neuronal cell type and maturational
stage (Calabrese et al. 2006; Sheng and Hoogenraad 2007). For example, the
adult mean number of dendritic spines on apical dendrites of pyramidal neurons
in layer III of visual cortex is 1 spine/µm,
versus 2-4 spines/µm in adult
hippocampal CA1 pyramidal and in granule cells, while several Purkinje cells
have more than ten spines/µm (Gould et al. 1990;
Fiala et al. 1998; Johnson et al. 2002). In these areas with more than one
synapse/µm, if each synaptobot possesses ten neuroelectrical
nanosensors (est. 3375 nm3 each (Martins et al. 2015)) then a single
robot can monitor action potentials on up to ten different synapses with
adequate temporal resolution. Assuming 2.42
x1014 synapses in the human brain (Martins et al. 2012) and with
each synaptobot servicing 1-10 synapses, the required number of 0.5 µm3 synaptobots is 24-242 x 1012
robots having total fleet volume of 1.2-12 x 1013 µm3 or 12-120 cm3, representing
~0.9-9% of total human brain volume. This lies within the 1-10% “safe” tissue and organ
intrusiveness limit recommended elsewhere (Freitas 2003).
5.6
Special nanorobot designs and protocols
Special
nanorobot designs and operational protocols may be required for areas in which
intracellular navigation is seriously constrained. Axon and dendritic regions
with less than 0.50 μm of diameter
are the most serious example (Shepherd and Harris 2010). The diameter of axons
within the mammalian nervous system had been reported to vary by a factor of
more than 100 (Liewald 2014), but closer study of the frequency distribution of
the diameters of myelinated axons in three different locations of the corpus
callosum in human brains suggested that 70-90 per cent of axon diameters are ≥0.50 μm, with minimum
and maximum diameters of 0.16 μm and 3.73 μm respectively, and a mean value and standard
deviation of 0.73 ± 0.55 μm (Liewald et al. 2014). A small percentage of 0.5 µm3 synaptobots might have difficulty
intracellularly reaching the most distal axonic and dendritic regions,
depending on nanorobot shape.
Special
robot-form factor design choices will be required for scanning structural
information inside some of the longest and thinnest dendritic spines. The
postsynaptic structure of 90 per cent of the excitatory synapses in the
mammalian brain are dendritic spines (Sheng and Hoogenraad 2007; Bhatt et al.
2009). These are functionally important because they can compartmentalize
calcium and serve a myriad of functions ranging from basic computational
operations to synaptic plasticity based on coincidence (Contreras 2004). Learning-associated structural changes involve
various synaptic changes including: (1) formation of new dendritic spines, (2)
changes in shape and size of existing dendritic spines, and (3) changes in the
organization of postsynaptic densities of spines (De Roo et al. 2008). Spine shapes (Figure 1) are commonly categorized as “mushroom” (~60%
of all spines in adult cortex), “thin” (10-20%,), “stubby” (20-25%,), “filopodia” (2-15%), and “branched” (a small number), with a continuum among these categories
(Sheng and Hoogenraad 2007). Across all types, ~0.5-2 µm diameter spine heads (Sheng and Hoogenraad 2007;
Sala et al. 2008) connect to the dendrite through 0.04-1 µm long necks (Calabrese et al. 2006). Most spines are
located on dendrites but some may also be present on the soma or the axonal
initial segment, and some spines receive more than one synapse, e.g., the
branched type (Spacek 2006).
Figure 1.Three dimensional
reconstruction of a hippocampal dendrite.
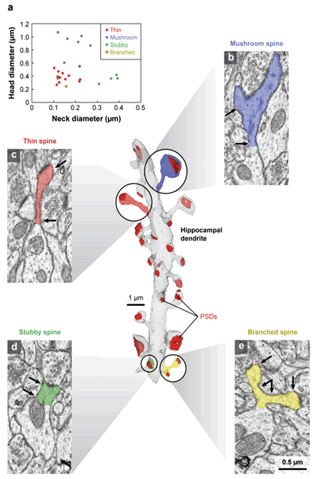
Three dimensional reconstruction of a hippocampal dendrite (gray) (from Area CA1 of the Rat) illustrate the four main types of different spine shapes, including mushroom (blue)(b), thin (red)(c), stubby (green)(d), and branched (yellow)(e). PSDs (red) also vary in size and shape. On (a) a graph plots the ratio of head diameters to neck diameters for the spines on the reconstructed dendrite. Arrows indicate where the head and neck diameters were measured for each spine in b–e. Scale bar = 0.5 μm. (Reprinted with Permission)(from (Bourne and Harris 2008)).
Synaptobots
should readily fulfill temporal resolution requirements for monitoring gross
changes in spine structure. Spine lifetimes vary greatly – stables ones may persist for at least a month, whereas
others may last for a few days or less (Trachtenberg 2002) – but spine shape can change over periods of minutes or
hours (Sala et al. 2008). Synaptobots should be able to distinguish pre-spinal
filopodia processes from regular synapses because filopodia have turnover times
measured in hours, with only ~20% of filopodia turning into spines and 80%
disappearing within 48 h (Alvarez and Sabatini 2007; Zuo et al. 2005).
Unilaterally
positioning sensors at either the pre- or the post-synaptic side of
axo-spinous, dendro-spinous, or soma-spinous synapses may be sufficient.
(Spino-spinous synapses, where two spines directly contact, are rare.) However,
for redundancy purposes it might be desirable for neuronanorobots to position
sensors on both synaptic sides. Such bilateral monitoring might be challenging
depending on spine morphology, diameter, and neck lengths. In especially narrow
cases, the mission design can include insertion of a nanomanipulator from the
opposite side.
Spine
monitoring includes not only volume and shape tracking but may also include
local organelle scanning when necessary. Spines typically have organelles
inside or near the entrance, including the smooth endoplasmic reticulum, spine
apparatus, actin cytoskeleton, endosomal compartments, translation machinery
components (e.g., polyribosomes), and the crucial postsynaptic density (PSD)
which occupies ~10 per cent of the surface area and is opposite to the
presynaptic active zone (Sheng and Hoogenraad 2007; Sala et al. 2008). Synaptic
plasticity is mediated by changes in the molecular composition of synaptic
proteins (Sheng and Hoogenraad 2007), and many of those proteins are located in
the PSD, a structure composed of ~1461 membranous and cytoplasmic proteins (Bayés 2010) localized at the postsynaptic plasma membrane
of excitatory synapses (Figure 2).
Figure
2: The variety of proteins in the PSD fraction.
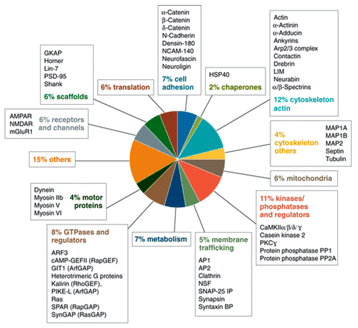
Illustration of some of the variety of
proteins on PSD (categorized according to cellular function). Only small
subsets of identified proteins are shown as examples (Reprinted with
Permission)(from (Sheng and Hoogenraad 2007)).
Nanorobotic monitoring
of the PSD seems essential. The PSD is a complex molecular machine that
dynamically changes its structure and composition in response to synaptic
activity, which dynamically regulates its components through protein
phosphorylation, palmitoylation, local protein translation, the
ubiquitin-proteasome system for protein degradation, and redistribution of
specific proteins, such as CaMKIIα and AMPARs, both to and away from the PSD (Kim and Ko 2006; Sheng and
Hoogenraad 2007). PSD proteins organize signaling pathways to coordinate
structural and functional changes in synapses. The PSD regulates trafficking
and recycling of glutamate receptors (which determines synaptic strength and
plasticity), promotes the formation and maturation of excitatory synapses by
co-aggregating with post-synaptic cell adhesion molecules, organizes
neurotransmitter receptors in the synaptic cleft, serves as a signaling
apparatus, and is an essential component of an extraordinary synaptic signaling
assemblage that has a strong relation with important mechanisms for synaptic
regulation, including long-term potentiation (LTP) and long-term depression
(LTD) (Sheng and Hoogenraad 2007).
The structure and
composition of the PSD is modified by external stimuli and by synaptic activity
over the time course of seconds to minutes and hours to days (Sheng and
Hoogenraad 2007). The “typical” PSD is a disklike structure with an average diameter
of 300-400 nm diameter (range 200-800 nm), a thickness of 30-60 nm (Baude et
al. 1993; Rácz et al. 2004; Okabe 2007; Sheng and Hoogenraad
2007), estimated volume 7.5×106
nm3 and mass ~1.1 GDa (Chen et al. 2005).
5.7 Measuring
synaptic plasticity
Nanorobots can directly
measure changes in synaptic plasticity. For instance, the activity-dependent
modification of PSD proteins over a timescale of seconds to hours is believed
to underlie plasticity processes such as LTP and LTD (Sheng and Hoogenraad
2007). Longer-term changes in PSD structure and composition (hours to days)
involve altered protein synthesis, either in the neuronal cell body or
dendrites (Sheng and Hoogenraad 2007). Degradation of PSD proteins by the
ubiquitin-proteasome system (Bingol and Schuman 2006) sculpts PSD structure and
plays a major role in synaptic plasticity. Remarkably, recent evidence even
points toward a rapid exchange of PSD proteins, such as AMPARs and PSD-95,
between neighboring synapses in steady-state conditions (Sheng and Hoogenraad
2007).
LTP and LTD events
cause structural changes to spines, altering spine number, size, shape, and
subcellular composition in both immature and mature spines (Bourne and Harris
2008). The spine neck acts as a diffusion barrier (controlled by neuronal
activity) to current flow and to the diffusion of molecules between the spine
head and the dendrite. The geometry of the spine neck determines calcium efflux
into the dendrite shaft and hence the degree of calcium elevation in the spine
head following NMDAR activation (Bloodgood and Sabatini 2005; Sheng and Hoogenraad
2007; Alvarez and Sabatini 2007).
In experimental work,
spines that received LTP induction increased in volume from 50-200 per cent
(Alvarez and Sabatini 2007) with the increase persisting for more than 1 hour
after stimulation (Alvarez and Sabatini 2007). LTP causes sustained spine head
enlargement due to F-actin polymerization, while LTD causes AMPA receptor
internalization with spine elongation and/or shrinkage of spine heads due to
actin depolymerization (Bourne and Harris 2008). More detailed synaptic
structural changes (within synaptic plasticity limits) occur in the:
(1) size and
composition of the postsynaptic density;
(2) assembly and
disassembly of actin filaments;
(3) exocytosis and
endocytosis of glutamate receptors and ion channels;
(4) regulation of local
protein synthesis by redistribution of polyribosomes and proteasomes;
(5) dynamic
repositioning of smooth endoplasmic reticulum (SER) and mitochondria; and
(6) metabolic and
structural interactions between spines and perisynaptic astroglia (Bourne and Harris 2008).
There is a clear strong
relation between synapse bouton size and shape, and the organelle and
macromolecular changes happening inside synapses. This provides some level of
redundancy of information, which suggests that monitoring all the spine
organelles and molecular components is very likely unnecessary. Synaptobots can
deduce a great deal of useful information when they scan the gross volume and
shape of the spine. The redundancy of information is expected to seriously reduce
the monitoring tasks of the synaptobots. However, to quantify and identify what
information is deducible from where will require more data on the range of
quantitative ultrastructural synaptic characteristics found throughout the
different brain areas. Details of the quantitative morphology of synapses are
still limited to a relatively small number of synapses (Rollenhagen and Lübke 2006), which points to the need for further
experimental data.
6. Conclusions
Fundamental,
life-saving, structural and functional human connectome-associated information
cannot be preserved using current destructive or non-destructive human brain
imaging technologies. Those technologies, as an extension of current form, seem
not to be theoretically scalable to the desired level of temporal or spatial
resolution required to preserve human cognitive life. Non-destructive
preservation of comprehensive in vivo whole-human brain information is
proposed to be achieved using a set of neuronanorobots, named endoneurobots,
gliabots, and synaptobots, conceptually designed in this paper. Such nanorobots
should preserve the underlying structural and functional information of the
human brain with appropriate temporal and spatial resolution.
Acknowledgments
The
principal author (NRBM) thanks the Fundação para a Ciência e Tecnologia (FCT) for their
financial support of this work (grant SFRH/BD/69660/2010). The author also
thanks for the support granted by Qualificar é Crescer, Quadro de Referência Estratégico Nacional, União Europeia Fundo Social Europeu, and
Governo da República
Portuguesa.
![]()
Glossary
of terms and abbreviations
Field-effect transistor (FET) is a transistor using an electric field
to control the electrical conductivity of a channel in a semiconductor
material.
Micro computed microtomography (Micro-CT) uses x-rays to
non-destructively create cross-section images of a certain physical object for
later 3D reconstruction.
Axon initial segment (AIS) is a specific axonic membrane region with
specific clusters of voltage-gated channels where action potentials are
frequently initiated.
The postsynaptic density (PSD) is a specific membrane region of a
postsynaptic neuron (in close apposition to the presynaptic active zone) being
particularly dense in proteins.
Magnetic resonance imaging (MRI) is a widely used medical imaging
technique using magnetic fields and radio waves to permit 3D anatomical and
physiological reconstructions of parts of the human body.
Functional magnetic resonance imaging (fMRI) is a neuro-imaging
technique for measuring brain activity by detecting blood flow associated
changes.
Resting state fMRI (R-fMRI) is a method for functional brain imaging of
the resting brain activity by detecting blood-oxygen-level dependent signals
measurable using functional magnetic resonance imaging.
Electron microscope (EM) is a microscope using beams of accelerated
electrons as its illumination source typically providing higher resolution than
light microscopy.
Serial block-face scanning electron microscopy (SBEM) is a method for
3D ultra high-resolution imaging using an ultramicrotome mounted inside a
vacuum chamber of a scanning electron microscope.
Transmission electron microscope (TEM) is a microscope using beams of
electrons transmitted through an ultra-thin specimen and focused onto a
sensorial layer, screen or device.
References
Alivisatos, A.P., M.
Chun, G.M. Church, R.J. Greenspan, M.L. Roukes, and R. Yuste. 2012. The Brain
Activity Map Project and the challenge of functional connectomics. Neuron 74(6):
970–74.
Allen, P. 2009. Piece of mind, the world in 2009. The Economist.
Allen Institute for
Brain Science. 2014. Allen Brain Atlas.
http://human.brain-map.org/ (accessed December 23, 2015).
Allen Institute for
Brain Science. 2014. Allen Institute for Brain Science. Allen Mouse Brain
Atlas.
http://mouse.brain-map.org/
(accessed December 23, 2015).
Alvarez, V.A., and B.L.
Sabatini. 2007. Anatomical and physiological plasticity of dendritic spines. Annual Reviews of Neuroscience 30: 79–97.
Amunts, K., C. Lepage,
L. Borgeat, H. Mohlberg, T. Dickscheid, M. Rousseau, S. Bludau, P. Bazin, L.B.
Lewis, A. Oros-Peusquens, N.J. Shah, T. Lippert, K. Zilles, and A.C. Evans.
2013. BigBrain: An ultrahigh-resolution 3D human brain model. Science 21 June. Vol. 340 no. 6139: 1472-1475.
Anderson, J., and
C.M.V. Itallie. 2009. Physiology and function of the tight junction. Cold
Spring Harbor Perspectives in Biology 1(2): a002584.
Anderson, J.R., B.W.
Jones, C.B. Watt, M.V. Shaw, J.H. Yang, D. Demill, J.S. Lauritzen, Y. Lin, K.D.
Rapp, D. Mastronarde, P. Koshevoy, B. Grimm, T. Tasdizen, R. Whitaker, and R.E.
Marc. 2011. Exploring the retinal connectome. Molecular Vision 17: 355–65.
Arking, R. 2006. Biology
of aging: Observations and principles. 3rd ed. New York: Oxford University
Press.
Ascoli. G. 2009. Coming of age of the hippocampome.
Front. Neur. Conference Abstract: Neuroinformatics 2009.
Astier, Y., H. Bayley,
and S. Howorka. 2005. Protein components for nanodevices. Current Opinion in Chemical Biology 9(6): 576–84.
Azevedo, F.A., L.R. Carvalho, L.T. Grinberg, J.M. Farfel, R.E. Ferretti,
R.E. Leite, W.J. Filho, R. Lent, and S. Herculano-Houzel. 2009. Equal numbers of neuronal and nonneuronal cells make
the human brain an isometrically scaled-up primate brain. Journal of
Comparative Neurology 513(5): 532–41.
Baude, A., Z. Nusser,
J.D. Roberts, E. Mulvihill, R.A. McIlhinney, and P. Somogyi. 1993. The
metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic
membrane of neuronal subpopulations as detected by immunogold reaction. Neuron
11(4): 771–87.
Bargmann, C.I., and E.
Marder. 2013. From the connectome to brain function. Nature Methods
10: 483–90.
Bayés, A. 2010. Characterization of the proteome, diseases
and evolution of the human postsynaptic density. Nature Neuroscience
14(1): 19–21.
Bertoni-Freddari, C., P
Fattoretti, T. Casoli, G. Di Stefano, M. Solazzi, E. Perna, and C. Angelis.
2006. Reactive structural dynamics of synaptic mitochondria in ischemic delayed
neuronal death. Annals of the New York
Academy of Sciences 1090(1): 26–34.
Bhatt, D.H., S. Zhang,
and W-B. Gan. 2009. Dendritic spine dynamics. Annual Review of Physiology
71: 261–82.
Bingol, B., and E.M.
Schuman. 2006. Activity-dependent dynamics and sequestration of proteasomes in
dendritic spines. Nature 441: 1144–1148.
Biswal, B.B., M. Mennes, X-N. Zuo, S. Gohel, C. Kelly, S.M.
Smith, C.F. Beckmann, J.S. Adelstein, R.L. Buckner, S. Colcombe, A-M.
Dogonowski, M. Ernst, D. Fair, M. Hampson, M.J. Hoptman, J.S. Hyde, V.J.
Kiviniemi, R. Kötter, S-J. Li, C-P- Lin, M.J. Lowe, C. Mackay, D.J.
Madden, K.H. Madsen, D.S. Margulies, H.S. Mayberg, K. McMahon, C.S. Monk, S.H.
Mostofsky, B.J. Nagel, J.J. Pekar, S.J. Peltier, S.E. Petersen, V. Riedl ,
S.A.R.B. Rombouts, B. Rypma, B.L. Schlaggar, S. Schmidt, R.D. Seidler, G.J.
Siegle, C. Sorg, G-J. Teng, J. Veijola, A. Villringer, M. Walter, L. Wang, X-C.
Weng, S. Whitfield-Gabrieli, P. Williamson, C. Windischberger, Y-F. Zang, H-Y.
Zhang, F.X. Castellanos, and M.P. Milham. 2010. Toward discovery science of
human brain function. Proceedings of
the National Academy of Sciences
107(10): 4734–4740.
Black, J.E., K.R.
Isaacs, B.J. Anderson, A.A. Alcantara, and W.T. Greenough. 1990. Learning
causes synaptogenesis, whereas motor activity causes angiogenesis, in
cerebellar cortex of adult rats. Proceedings
of the National Academy of Sciences
87: 5568–5572.
Bliss, T.V.P., and G.L. Collingridge. 1993. A synaptic model of
memory: Long-term potentiation in the hippocampus. Nature 361: 31–39.
Bloodgood, B.L., and
B.L. Sabatini. 2005. Neuronal activity regulates diffusion across the neck of
dendritic spines. Science 4: 866–69.
Bock, D.D., W-C.A. Lee,
A.M. Kerlin, M.L. Andermann, G. Hood, A.W. Wetzel, S. Yurgenson, E.R. Soucy,
H.S. Kim, and R.C. Reid. 2011. Network anatomy and in vivo
physiology of visual cortical neurons. Nature 471: 177–82.
Bohland, C.W., H.
Barbas, H. Bokil, M. Bota, H.C. Breiter, H.T. Cline, J.C. Doyle, P.J. Freed,
R.J. Greenspan, S.N. Haber, M. Hawrylycz, D.G. Herrera, C.C. Hilgetag, Z.J.
Huang, A. Jones, E.G. Jones, H.J. Karten, D. Kleinfeld, R. Kötter, H.A. Lester, J.M. Lin, B.D. Mensh, S. Mikula, J.
Panksepp, J.L. Price, J. Safdieh, C.B. Saper, N.D. Schiff, J.D. Schmahmann,
B.W. Stillman, K. Svoboda, L.W. Swanson, A.W. Toga, D.C. Van Essen, J.D.
Watson, and P.P. Mitra. 2009. A proposal for a coordinated effort for the
determination of brainwide neuroanatomical connectivity in model organisms at a
mesoscopic scale. PLoS Computational Biology 5(3): e1000334.
Bourne, J.N., and K.M.
Harris. 2008. Balancing structure and function at hippocampal dendritic spines.
Annual Review of Neuroscience 31: 47–67.
Briggman, K.L., and
D.D. Bock. 2012. Volume electron microscopy for neuronal circuit
reconstruction. Current Opinion in
Neurobiology 22: 154–61.
Bumbarger, D. J., M. Riebesell, C. Rödelsperger, and R.J. Sommer. 2013. System-wide
rewiring underlies behavioral differences in predatory and bacterial-feeding
mematodes. Cell 152(1): 109–119.
Calabrese B., M.S.
Wilsos, and S. Halpain. 2006. Development and regulation of dendritic spine
synapses. Physiology 21(1): 38–47.
Chen, X., L. Vinade,
R.D. Leapman, J.D. Petersen, T. Nakagawa, T.M. Phillips, M. Sheng, and T.S.
Reese. 2005. Mass of the postsynaptic density and enumeration of three key
molecules. Proceedings of the National
Academy of Sciences 102: 11551–11556.
Chen, B.L., D.H. Hall,
and D.B. Chklovskii. 2006. Wiring optimization can relate neuronal structure
and function. Proceedings of the
National Academy of Sciences 103:
4723–4728.
Chung, K. 2013.
Structural and molecular interrogation of intact biological systems. Nature 497:
332–37.
Chung, K., and K.
Deisseroth. 2013. CLARITY for mapping the nervous system. Nature Methods
10(6): 508-13.
Clayden, J.D. 2013.
Imaging connectivity: MRI and the structural networks of the brain. Functional Neurology 28: 197–203.
Connectome Project.
2011. Open connectome project: Collectively reverse engineering the brain, one
synapse at a time.
http://www.openconnectomeproject.org (accessed December 23, 2015).
Contreras, D. 2004.
Electrophysiological classes of neocortical neurons. Neural Networks 17(5–6): 633–46.
Craddock, R.C., S.
Jbabdi, C.G. Yan, J.T. Vogelstein, F.X. Castellanos, A. Di Martino, C. Kelly,
K. Heberlein, S. Colcombe, and M.P. Milham. 2013. Imaging human connectomes at
the macroscale. Nature Methods 10: 524–39.
De Roo, M., P. Klauser,
P.M. Garcia, L. Poglia, and D. Muller. 2008. Spine dynamics and synapse
remodeling during LTP and memory processes. Progress in Brain Research 169:
199–207.
Deco, G., A. Ponce-Alvarez, D. Mantini, G.L. Romani, P.
Hagmann, and M. Corbetta. 2013. Resting-state functional connectivity emerges
from structurally and dynamically shaped slow linear fluctuations. Journal of Neuroscience 33(27): 11239 –11252.
DeFelipe, J., and I.
Fariñas. 1992. The pyramidal neuron of the cerebral cortex:
Morphological and chemical characteristics of the synaptic inputs. Progress
in Neurobiology 39: 563–607.
Dekosky, S.T., and S.
Scheff. 1990. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Annals
of Neurology 27(5): 457–64.
Donohue, D.E., and G.A.
Ascoli. 2008. A comparative computer simulation of dendritic morphology. PLoS
Computational Biology 4(6): e1000089.
FCP. 2010. 1000
Functional Connectomes Project.
http://fcon_1000.projects.nitrc.org/fcpClassic/FcpTable.html (accessed January 5, 2196).
Fiala, J.C., M.
Feinberg, V. Popov, and K.M. Harris. 1998. Synaptogenesis via dendritic
filopodia in developing hippocampal area CA1. Journal of Neuroscience
18: 8900–8911.
Fiala, J.C., and K.M. Harris. 2001. Extending
unbiased stereology of brain ultrastructure to three-dimensional volumes. Journal
of the American Medical Informatics Association 8(1): 1–16.
Freitas, R.A., Jr. 1999. Nanomedicine.
Volume I: Basic capabilities. Georgetown, TX: Landes Bioscience.
Freitas, R.A., Jr. 2000. Nanodentistry. Journal of the American Dental
Association 131: 1559–1566.
Freitas, R.A., Jr. 2003. Nanomedicine.
Volume IIA: Biocompatibility. Georgetown, TX: Landes Bioscience.
Freitas, R.A., Jr. 2005a. What is
nanomedicine? Disease-A-Month 51: 325–41.
Freitas, R.A., Jr. 2005b. Current
status of nanomedicine and medical nanorobotics. Journal of Computational
and Theoretical Nanoscience 2: 1–25.
Freitas, R.A., Jr. 2005c.
Microbivores: Artificial mechanical phagocytes using digest and discharge
protocol. Journal of Evolution and Technology 14 (April): 1–52.
Available online http://jetpress.org/volume14/freitas.pdf (accessed December 23, 2015).
Freitas, R.A., Jr. 2006a.
Pharmacytes: An ideal vehicle for targeted drug delivery. Journal of
Nanoscience and Nanotechnology 6: 2769–2775.
Freitas, R.A., Jr.
2006b. Progress in nanomedicine and medical nanorobotics. In Handbook of
theoretical and computational nanotechnology. Volume 6: Bioinformatics,
nanomedicine, and drug design, ed. M. Rieth, and W. Schommers, 619–72. Stevenson Ranch, CA: American Scientific
Publishers.
Freitas, R.A., Jr. 2007. The ideal
gene delivery vector: Chromallocytes, cell repair nanorobots for chromosome
replacement therapy. Journal of Evolution and Technology 16 (June): 1–97.
Available online http://jetpress.org/v16/freitas.pdf (accessed December 23, 2015).
Freitas, R.A., Jr. 2010.
Comprehensive nanorobotic control of human morbidity and aging. In The
future of aging: Pathways to human life extension, ed. G.M. Fahy, M.D.
West, L.S. Coles, and S.B. Harris, 685–805. New York: Springer.
Friston, K.J. 2011.
Functional and effective connectivity: A review. Brain Connectivity 1: 13–36.
Fuhrmann, G., I. Segev,
H. Markram, and M. Tsodyks. 2002. Coding of temporal information by
activity-dependent synapses. Journal of Neurophysiology 87(1): 140–48.
GE-MCS. 2013. Phoenix
Nanotom M.
http://www.ge-mcs.com/en/radiography-x-ray/ct-computed-tomography/nanotom-s.html
or
https://www.gemeasurement.com/sites/gemc.dev/files/nanotom_brochure_english_0.pdf (accessed December 23, 2015).
Gross, C.G. 2000.
Neurogenesis in the adult brain: Death of a dogma. Nature Reviews
Neuroscience 1: 67–73.
Gould, E., C.S.
Woolley, M. Frankfurt, and B.S. McEwen. 1990. Gonadal steroids regulate
dendritic spine density in hippocampal pyramidal cells in adulthood. Journal
of Neuroscience 10: 1286–1291.
Harris, K. M. 1999.
Synapse Web.
http://synapses.clm.utexas.edu (accessed December 23, 2015).
Hayworth, K.J., N.
Kasthuri, R. Schalek, and J.W. Lichtman. 2006. Automating the collection of
ultrathin serial sections for large volume TEM reconstructions. Microscopy and Microanalysis 12(Suppl 2): 86–87.
Hayworth, K.J. (2012)
Electron imaging technology for whole brain neural circuit mapping. International Journal of Machine Consciousness 4(1) 87–108.
Helmholtz Association.
2010. New microscope reveals ultrastructure of cells. Public release: Nov 19.
Helmstaedter, M., K.L.
Briggman, and W. Denk. 2008. 3D structural imaging of the brain with photons and
electrons. Current Opinion in
Neurobiology 18: 633–41.
Helmstaedter, M., K.L. Briggman, and W. Denk. 2011. High-accuracy
neurite reconstruction for high-throughput neuroanatomy. Nature Neuroscience 14: 1081–1088.
Herculano-Houzel,
S. 2009. The human brain in numbers: A linearly scaled-up primate brain. Frontiers in Human Neuroscience 9(3): 31.
Holtmaat, A., and K.
Svoboda. 2009. Experience-dependent structural synaptic plasticity in the
mammalian brain. Nature Reviews Neuroscience 10: 647–658.
Human Brain Project.
2013. Human Brain Project.
https://www.humanbrainproject.eu (accessed
January 5, 2016).
IBM. 2008. IBM seeks to
build the computer of the future based on insights from the brain – IBM awarded DARPA funding for cognitive computing
collaboration.
https://www-03.ibm.com/press/us/en/pressrelease/26123.wss (accessed
December 23, 2015).
IHGSC. 2004.
International Human Genome Sequencing Consortium: Finishing the euchromatic
sequence of the human genome. Nature 431: 931–45.
Ingber, D.E. 2006.
Cellular mechanotransduction: Putting all the pieces together again. Journal
of the Federation of American Societies for Experimental Biologies (FASEB) 20(7):
811–27.
Islam, S. 2011.
Characterization of the single-cell transcriptional landscape by highly
multiplex RNA-seq. Genome Research 21: 1160–1167.
Izquierdo, E.J., and
R.D. Beer 2013. Connecting a connectome to behavior: An ensemble of
neuroanatomical models of C. elegans klinotaxis. PLoS Computational Biology
9(2): e1002890.
Johnson, G.A. 2010. An
image-based reference for coordinating mouse brain research. NeuroImage
53(2): 365.
Johnson,
M.H., Y. Munakata, and R. Gilmore. 2002. Brain development and cognition: A reader.
2nd Edition. Oxford: Blackwell.
Jones, A. 2011. Allen
Institute: A map of the brain, TED Global Talk. Weblink: https://www.ted.com/talks/allan_jones_a_map_of_the_brain?language=en (accessed December 23, 2015)
Jones, E.G., and L.M.
Mendell. 1999. Assessing the decade of the brain. Science
284(5415): 739.
Kandel, E.R., J. Schwartz, and T. Jessell. 1991. Principles of
neural science. 3rd ed. New York: McGraw-Hill.
Kandel, E.R. 2001. The
molecular biology of memory storage: A dialogue between gene and synapses. Science
294 (5544): 1030–1038.
Kandel, E.R., J.
Schwartz, and T. Jessell. 2000. Principles
of neural science. 4th ed. New York:
McGraw Hill.
Kandel, E.R., H. Markram, P.M. Matthews, R. Yuste, and C.
Koch. 2013. Neuroscience thinks big (and collaboratively). Nature Reviews Neuroscience 14: 659–64.
Kasthuri, N., K.
Hayworth, J.C. Tapia, R. Schalek, S. Nundy, and J.W. Lichtman. 2009. Thin
section scanning electron microscopy of serial sections of brain on tape. Neuroscience Meeting Planner. Chicago, IL: Society for
Neuroscience, 2009.
Kasthuri, N., and J.W.
Lichtman. 2007. The rise of the “projectome.” Nature Methods 4(4):
307–308.
Kim, E., and J. Ko.
2006. Molecular organization and assembly of the postsynaptic density of excitatory
brain synapses. Results and Problems in Cell Differentiation 43: 1–23.
Kleinfeld, D.,
A. Bharioke, P. Blinder, D.D. Bock, K.L. Briggman, D.B. Chklovskii, W. Denk, M.
Helmstaedter, J.P. Kaufhold, W-C. A. Lee, H.S. Meyer, K.D. Micheva, M. Oberlaender,
S. Prohaska, R.C. Reid, S.J. Smith, S. Takemura, P.S. Tsai, and B. Sakmann.
2011. Large-scale automated histology in the pursuit of connectomes. Journal
of Neuroscience 31(45): 16, 125–16, 138.
Kole, M.H. P., J.J.
Letzkus, and G.J. Stuart. 2007. Axon initial segment Kv1 channels control
axonal action potential waveform and synaptic efficacy. Neuron 55(4):
633–47.
Kostarelos K. 2010.
Nanorobots for medicine: How close are we? Nanomedicine 5(3): 341–42.
Kreutzberg, G. W. 1995.
The first line of defense in brain pathologies. Drug Research 45(1): 357–60.
Kurzweil, R. 2005. The
Singularity is near. New York: Viking.
Lee, S.H., J.H. Choi, N. Lee, H.R. Lee, J.L. Kim, N.K. Yu, S.L.
Choi, S.H. Lee, H. Kim, and B.K. Kaang. 2008. Synaptic protein degradation
underlies destabilization of retrieved fear memory. Science 319(5867):
1253–1256.
Lein, E.S., M.J.
Hawrylycz, N. Ao, M. Ayres, A. Bensinger, A. Bernard, A.F. Boe, M.S. Boguski,
K.S. Brockway, E.J. Byrnes, L. Chen, L. Chen, T-M. Chen, M.C. Chin, J. Chong,
B.E. Crook, A. Czaplinska, C.N. Dang, S. Datta, N.R. Dee, A.L. Desaki, T.
Desta, E. Diep, T.A. Dolbeare, M.J. Donelan, H-W. Dong, J.G. Dougherty, B.J.
Duncan, A.J. Ebbert, G. Eichele, L.K. Estin, C. Faber, B.A. Facer, R. Fields,
S.R. Fischer, T.P. Fliss, C. Frensley, S.N. Gates, K.J. Glattfelder, K.R.
Halverson, M.R. Hart, J.G. Hohmann, M.P. Howell, D.P. Jeung, R.A. Johnson, P.T.
Karr, R. Kawal, J.M. Kidney, R.H. Knapik, C.L. Kuan, J.H. Lake, A.R. Laramee,
K.D. Larsen, C. Lau, T.A. Lemon, A.J. Liang, Y. Liu, L.T. Luong, J. Michaels,
J.J. Morgan, R.J. Morgan, M.T. Mortrud, N.F. Mosqueda, L.L. Ng, R. Ng, G.J.
Orta, C.C. Overly, T.H. Pak, S.E. Parry, S.D. Pathak, O.C. Pearson, R.B.
Puchalski, Z.L. Riley, H.R. Rockett, S.A. Rowland, J.J. Royall, M.J. Ruiz, N.R.
Sarno, K. Schaffnit, N.V. Shapovalova, T. Sivisay, C.R. Slaughterbeck, S.C.
Smith, K.A. Smith, B.I. Smith, A.J. Sodt, N.N. Stewart, K-R. Stumpf, S.M.
Sunkin, M. Sutram, A. Tam, C.D. Teemer, C. Thaller, C.L. Thompson, L.R. Varnam,
A. Visel, R.M. Whitlock, P.E. Wohnoutka, C.K. Wolkey, V.Y. Wong, M. Wood, M.B.
Yaylaoglu, R.C. Young, B.L. Youngstrom, X.F. Yuan, B. Zhang, T.A. Zwingman, and
A.R. Jones. 2007. Genome-wide atlas of gene expression in the adult mouse
brain. Nature 445: 168–76.
Lelièvre, S. A. 2009. Contributions of extracellular matrix
signaling and tissue architecture to
nuclear mechanisms and spatial organization of gene expression control. Biochimica et Biophysica Acta 1790(9): 925–35.
Lichtman, J., J. Livet,
and J.R. Sanes. 2008. A technicolour approach to the connectome. Nature Reviews Neuroscience 9(6): 417–22.
Liewald, D., R. Miller,
N. Logothetis, H.J. Wagner, and A. Schüz.
2014. Distribution of axon diameters in cortical white matter: An electron-microscopic
study on three human brains and a macaque. Biological Cybernetics
108(5): 541–57.
Liu, X. 2012.
Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature
484: 381–85.
Livet J., T.A.
Weissman1, H. Kang, R.W. Draft, J. Lu, R.A. Bennis, J.R. Sanes, and J.W.
Lichtman. 2007. Transgenic strategies for combinatorial expression of
fluorescent proteins in the nervous system. Nature 450: 56–62.
Lu,
J.J.C. Tapia, O.L. White, and J.W. Lichtman. 2009. The interscutularis muscle
connectome. PLoS Biology 7(4): e1000108.
MacAskill, A.F., J.E.
Rinholm, A.E. Twelvetrees, I.L. Arancibia-Carcamo, J. Muir, A. Fransson, P.
Aspenstrom, D. Attwell, and J.T. Kittler. 2009. Miro1 is a calcium sensor for
glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61:
541–55.
Mallouk, T. E., and A.
Sen. 2009. Powering nanorobots. Scientific
American 300: 72–77.
Ramaswami, S. J-D.
Courcol, M. Abdellah, S.R. Adaszewski, N. Antille, S. Arsever, G. Atenekeng, A.
Bilgili, Y. Brukau, A. Chalimourda, G. Chindemi, F. Delalondre, R. Dumusc, S.
Eilemann, M.E. Gevaert, P. Gleeson, J.W. Graham, J.B. Hernando, L. Kanari, Y.
Katkov, D. Keller, J.G. King, R. Ranjan, M.W. Reimann, C. Rössert, Y. Shi, J. C. Shillcock, M. Telefont, W.V.
Geit, J.V. Diaz, R. Walker, Y. Wang, S.M. Zaninetta, J. DeFelipe, S.L. Hill, J.
Muller, I. Segev, F. Schürmann, E.B. Muller,
and H. Markram. 2015. The neocortical microcircuit collaboration portal: A
resource for rat somatosensory cortex. Frontiers in Neural Circuits Vol
9, Article 44.
Markram, H. 2006. The
Blue Brain Project. Nature Reviews Neuroscience 7: 153–160.
Martel, S., M.
Mohammadi, O. Felfoul, Z. Lu, and P. Pouponneau. 2009. Flagellated
magnetotactic bacteria as controlled MRI-trackable propulsion and steering
systems for medical nanorobots operating in the human microvasculature. International Journal of Robotics Research 28(4): 571–82.
Martins, N.R.B., W.
Erlhagen, and R.A. Freitas, Jr. 2012. Non-destructive whole-brain monitoring
using nanorobots: Neural electrical data rate requirements. International Journal of Machine Consciousness 4: 109–140.
Martins, N.R.B., W.
Erlhagen, and R.A. Freitas, Jr. 2015. Action potential monitoring using
neuronanorobots: Neuroelectric nanosensors. International Journal of
Nanomaterials and Nanostructures 1: 20–41.
Mavroides, D., and A.
Ferreira, eds. 2011. NanoRobotics:
Current approaches and techniques.
New York: Springer.
McAllister, A.K. 2007.
Dynamic aspects of CNS synapse formation. Annual Review of Neuroscience
30: 425–50.
Miyamoto, K. 1986.
Numerical density estimation of mitochondria in thick slice by transmission
emission microscopy. Science on form: Proceedings of the First
International Symposium for Science on Form, 517–25. Tokyo: KTK Scientific Publishers.
Modha-blog 2009. A
proposal for mouse connectivity project.
http://p9.hostingprod.com/@modha.org/blog/2009/01/a_proposal_for_mouse_connectiv.html
(accessed December 23,
2015).
Mori, S., and J. Zhang.
2006. Principles of diffusion tensor imaging and its applications to basic
neuroscience research. Neuron 51: 527–39.
Morris, K. 2001.
Macrodoctor, come meet the nanodoctors. Lancet.
357(9258): 778.
Morris, R.L., and P.J.
Hollenbeck. 1995. Axonal transport of mitochondria along microtubules and
F-actin in living vertebrate neurons. Journal
of Cell Biology 131: 1315–1326.
Newman, E.A. 2001.
Propagation of intercellular calcium waves in retinal astrocytes and Müller cells. Journal of Neuroscience 21(7): 2215–2223.
NIH News. 2009. NIH
Launches the Human Connectome Project to unravel the brain’s connections. Press release, July 16.
Nopoulos, P., M. Flaum,
D. O’Leary, and N.C. Andreasen. 2000. Sexual dimorphism in
the human brain: Evaluation of tissue volume, tissue composition and surface
anatomy using magnetic resonance imaging. Psychiatry Research: Neuroimaging
98(1): 1–13.
Okabe S. 2007.
Molecular anatomy of the postsynaptic density. Molecular and Cellular
Neuroscience 34(4): 503–518.
Pakkenberg, B., and
H.J.G. Gundersen. 1997. Neocortical neuron number in humans: Effect of sex and
age. Journal of Comparative Neurology 384: 312–320.
Palmer, L. M., and G.
J. Stuart. 2006. Site of action potential initiation in Layer 5 pyramidal
neurons. Journal of Neuroscience 26(6): 1854–1863.
Park, H.H., A.C.
Jamison, and T.R. Lee. 2007. Rise of the nanomachine: The evolution of a
revolution in medicine. Nanomedicine 2(4): 425–39.
Patel, G.M., G.C.
Patel, R.B. Patel, J.K. Patel, and M. Patel. 2006. Nanorobot: A versatile tool
in nanomedicine. Journal of Drug
Targeting 14(2): 63–67.
Perrot, R., R. Berges,
A. Bocquet, and J. Eyer. 2008. Review of the multiple aspects of neurofilament
functions, and their possible contribution to neurodegeneration. Molecular
Neurobiology 38(1): 27–65.
Popov, A.M., Y. E
Lozovik, S. Fiorito, and L’H. Yahia. 2007.
Biocompatibility and applications of carbon nanotubes in medical nanorobots. International Journal of Nanomedicine 2(3): 361–72.
Qu, L., Y. Akbergenova,
Y. Hu, and T. Schikorski. 2009. Synapse-to-synapse variation in mean synaptic
vesicle size and its relationship with synaptic morphology and function. Journal
of Comparative Neurology 514(4): 343–52.
Rácz, B., T.A. Blanpied, M.D. Ehlers, and R.J. Weinberg.
2004. Lateral organization of endocytic machinery in dendritic spines. Nature
Neuroscience 7: 917–18.
Rengachary, S.S., and
R.G., Ellenbogen, eds. 2005. Principles
of neurosurgery. Amsterdam: Mosby-Elsevier.
Rollenhagen,
A.,and J.H.R. Lübke. 2006. The morphology of excitatory central synapses: From structure
to function. Cell Tissue Res 326: 221–37.
Rollenhagen, A., K. Sätzler, E.P. Rodríguez, P. Jonas, M. Frotscher, and J.H. Lübke. 2007. Structural determinants of transmission at large
hippocampal mossy fiber synapses. Journal of Neuroscience 27(39): 10434–10444.
Sabatini, B.L., and W.G. Regehr. 1996.Timing
of neurotransmission at fast synapses in the mammalian brain. Nature
384: 170–72.
Sala, C., I. Cambianica, and F. Rossi. 2008. Molecular
mechanisms of dendritic spine development and maintenance. Acta
Neurobiologiae Experimentalis 68(2): 289–304.
Sandberg, A., and N.
Bostrom. 2008. Whole brain emulation: A roadmap. Technical report. Future of
Humanity Institute, Oxford University.
Scheff, S.W., and D.A.
Price. 2006. Alzheimer’s disease-related
alterations in synaptic density: Neocortex and hippocampus. Journal of
Alzheimer's Disease 9(3 Suppl): 101–115.
Schwarz, T.L. 2013.
Mitochondrial trafficking in neurons. Cold
Spring Harbor Perspectives in Biology
5(6): 5:a011304.
Seung, H.S. 2010. TED
Talks – Sebastian Seung: I am my connectome. July, 2010.
Oxford, England.
Seung, H.S. 2011.
Neuroscience: Towards functional connectomics. Nature 471: 170–72.
Sheng, M., and C.C.
Hoogenraad. 2007. The postsynaptic architecture of excitatory synapses: A more
quantitative view. Annual Review of Biochemistry 76: 823–47.
Sheng, Z. H. 2014.
Mitochondrial trafficking and anchoring in neurons: New insight and
implications. Journal of Cell Biology 204( 7): 1087–1098.
Shepherd, G.M.G., and
K.M. Harris. 2010. Three-dimensional structure and composition of CA33CA1 axons
in rat hippocampal slices: Implications for presynaptic connectivity and
compartmentalization. Journal of
Neuroscience 18(20): 8300–8310.
Shin, S.S., T.
Verstynen, S. Pathak, K. Jarbo, A.J. Hricik, M. Maserati, S.R. Beers, A.M.
Puccio, F.E. Boada, D.O. Okonkwo, and W. Schneider. 2012. High-definition fiber
tracking for assessment of neurological deficit in a case of traumatic brain
injury: Finding, visualizing, and interpreting small sites of damage: Case
report video. Journal of Neurosurgery 116(5): 1062–1069.
Smith, S.M., R. Renden,
and H. von Gersdorff. 2008. Synaptic vesicle endocytosis: Fast and slow modes
of membrane retrieval. Trends in Neurosciences 31(11): 559–68.
Spacek, J. 2006.
Comparative morphology of dendritic spines and spine synapses. From: Synapse
Web.
Spacek, J., and K.
Sorra. 2007. Synapses of stratum radiatum. Synapse.
http://synapses.clm.utexas.edu/anatomy/radiatum/synapses.stm (accessed December 23, 2015).
Sporns, O., G. Tononi,
and R. Kötter. 2005. The human connectome: A structural
description of the human brain. PLoS Computational Biology 1(4): e42.
Südhof, T.C. 2004. The synaptic vesicle cycle. Annual
Review of Neuroscience 27: 509–547.
Sun, T., H. Qiao, P-Y.
Pan, Y. Chen, and Z-H. Sheng. 2013. Motile axonal mitochondria contribute to
the variability of presynaptic strength. Cell Reports 4: 413–419.
Syková, E., and C. Nicholson. 2008. Diffusion in brain
extracellular space. Physiology Reviews 88: 1277–1340.
Terry, R.D., E. Masliah, D.P. Salmon, N. Butters, R.
DeTeresa, R. Hill, L.A. Hansen, and R. Katzman. 1991. Physical basis of
cognitive alterations in Alzheimer’s
disease: Synapse loss is the major correlate of cognitive impairment. Annals
of Neurology 30(4): 572–80.
Theodore, T.W., R.
Preissl, P. Datta, M. Flickner, R. Singh, S.K. Esser, E. McQuinn, R. Appuswamy,
W.P. Risk, H.D. Simon, and D.S. Modha. 2012. 10^14, IBM Research Report.
Weblink: http://domino.research.ibm.com/library/cyberdig.nsf/papers/19B9020D53E753DB85257AB7005FFA18/$File/RJ10502.pdf (accessed December 23, 2015)
Thorne, R.G., and C.
Nicholson. 2006. In vivo diffusion analysis with quantum dots and
dextrans predicts the width of brain extracellular space. Proceedings of the
National Academy of Sciences 103: 5567–5572.
Tkachuk, A., F. Duewer,
H. Cui, M. Feser, S. Wang, and W. Yun. 2007. X-ray computed tomography in
Zernike phase contrast mode at 8 keV with 50-nm resolution using Cu rotation
anode X-ray source. Zeitschrift für Kristallographie 222(11): 650–55.
Trachtenberg, J.T.
2002. Long-term in vivo imaging of experience-dependent synaptic
plasticity in adult cortex. Nature 420: 788–94.
Travis E. R., and R. M.
Wightman. 1998. Spatio-temporal resolution of exocytosis from individual cells.
Annual Review of Biophysics and Biomolecular Structure 27: 77–103.
Trushina, E., E.
Nemutlu, S. Zhang, T. Christensen, J. Camp, J. Mesa, A. Siddiqui, Y. Tamura, H.
Sesaki, T.M. Wengenack, P.P. Dzeja, and J.F. Poduslo. 2012. Defects in
mitochondrial dynamics and metabolomic signatures of evolving energetic stress
in mouse models of familial Alzheimer’s
disease. PLoS ONE 7(2): e32737.
Vaidyanathana, M., L.P.
Clarke, C. Heidtman, R.P. Velthuizen, and L.O. Hall. 1997. Normal brain volume
measurements using multispectral MRI segmentation. Magnetic Resonance
Imaging 15(1): 87–97.
Varshney, C., B.L.
Chen, E. Paniagua, D.H. Hall, and D.B. Chklovskii. 2011. Structural properties
of the C. elegans neuronal network. PLoS Computational Biology 3(7):
e1001066.
White, J.G., E.
Southgate, J.N. Thomson, and S. Brenner. 1986. The structure of the nervous
system of the nematode Caenorhabditis elegans. Philosophical Transactions of
the Royal Society B: Biological Sciences 314: 1–340.
Wormatlas. 2008.
Wormatlas: A database featuring behavioral and structural anatomy of Caenorhabditis
elegans. http://wormatlas.org/ (accessed December 23, 2015).
Zador, A., J. Dubnau, H.K. Oyibo, H. Zhan, G. Cao, and
I.D. Peikon. 2012. Sequencing the connectome. PLoS Biology 10(10):
e1001411.
Zuo, Y., A. Lin, P. Chang, and W-B. Gan. 2005. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46(2): 181–89.