|
A peer-reviewed electronic journal published by the Institute for Ethics and ISSN 1541-0099 Vol. 16 – June 2007 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
The Ideal Gene Delivery Vector: Chromallocytes, Cell Repair Nanorobots for Chromosome Replacement Therapy
Robert A. Freitas Jr.
Journal of Evolution and Technology
- Vol. 16 Issue 1 - June 2007 - pgs 1-97
Outline of the Paper 1. Introduction 2. Basic Structure of the Cell Nucleus 2.1 Nuclear Envelope 2.2 Nuclear Interior, Chromosomes and DNA 2.3 Nucleolus 3. Chromallocyte Structure and Function 3.1 Overall Nanorobot Structure 3.2 Proboscis Manipulator 3.3 Funnel Assembly 3.4 Chromatin Storage Vaults 3.5 Mobility System 3.6 Power Supply 3.7 Onboard Computers 3.8 Summary of Primary and Support Subsystem Scaling 4. Ex Vivo Chromosome Sequencing and Manufacturing Facility 4.1 Genome Sampling and Modification 4.2 Chromosome Sequencing 4.3 Chromatin Synthesis 5. Mission Description 5.1 Mission Summary 5.2 Detailed Sequence of Chromallocyte Activities 6. Special Cases and Alternate Missions 6.1 Proliferating Cells 6.2 Pathological Cells 6.3 Brain, Bone, and Mobile Cells 6.4 Multinucleate Cells 6.5 Karyolobism and Karyomegaly 6.6 Mitochondrial DNA 6.7 Nonpathological Mosaicism 6.8 Partial- or Single-Chromosome CRT 6.9 Single-Cell and Whole-Body CRT 6.10 Heteroiatrogeny 6.11 Nanorobot Malfunction 7. Conclusions Acknowledgments References
1. Introduction Most human diseases involve a molecular malfunction at the cellular level, and cell function is largely controlled by gene expression and its resulting protein synthesis. As a result, many disease processes are driven either by defective chromosomes [1] or by defective gene expression [2]. One common practice of genetic therapy which has enjoyed only limited success is to supplement existing genetic material by inserting new genetic material into the cell nucleus, commonly using viral [3-5], bacteriophage [6], bacterial [7], stem cell [8], plasmid/phospholipid microbubble [9], cationic liposome [10], dendrimeric [11], chemical [12, 13], nanoparticulate [14, 15] or other appropriate transfer vectors to breach the cell membrane. However, permanent gene replacement using viral carriers has largely failed thus far in human patients due to immune responses to antigens of the viral carrier [16] as well as inflammatory responses, insertional mutagenesis, and transient effectiveness. Excess gene copies [17-19], repeat gene clusters [20], and partial trisomies [21] and higher polysomies [22] can often cause significant pathologies, sometimes mimicking aging [23]. Attempting to correct excessive expression caused by these errors by implementing antisense transcription silencing [24] on a whole-body, multi-gene, or whole-chromosome basis would be far less desirable than developing more effective therapeutic methods that did not require such extensive remediation. Electroporation [25] is another classic technique that uses electrical pulses to render cell membranes temporarily permeable to DNA, but this method cannot target individual cells in vivo and transfer is not perfect. Nucleofection [26] is a variant of electroporation that permits direct transfer of DNA into the nucleus, but only for in vitro applications. Lasers have been used to usher DNA, even sperm, into cells: using nanosecond UV pulses, some DNA is transferred, but the cells may be damaged irreparably. Femtosecond near-IR pulses greatly reduce cell damage [27] but DNA uptake is still seriously limited in scope and reliability. Mechanical injection into tissues of naked DNA plasmids carrying human cDNA into cells has shown promise [28], but only small lengths of DNA can be transferred and expressed in this manner. Direct microsurgical extraction of chromosomes from nuclei has been practiced since the 1970s [29-32], and microinjection of new DNA directly into the cell nuclei using a micropipette (pronuclear microinjection) is a common biotechnology procedure [33] easily survived by the cell, though such injected DNA often eventually exits the nucleus [34]. The commercial practice of DNA microinjection into pronuclei of zygotes from various farm animal species since 1985 has also shown poor efficiency and involves a random integration process which may cause mosaicism, insertional mutations and varying expression due to position effects [35]. Finally, for more than four decades microbiologists have used nuclear transfer [36] and nuclear transplantation [37] techniques to routinely extract or insert an entire nucleus into an enucleated cell using micropipettes without compromising cell viability, but such direct manual transfer approaches are impractical for in vivo therapeutic use in diseased tissues comprising billions or trillions of individual cells. Nuclear reprogramming [38] employs global resetting of epigenetic modifications only, without direct changes to nuclear DNA information. Purposeful intracellular infection by engineered bacteria containing desired supplementary genetic material might also be possible, given the presence of multiple endosymbionts with integrated genomes in some natural species [38a], but this biotechnology has not yet been developed. Nanomedicine and medical nanorobotics [39, 40] offers the prospect of powerful new tools for the treatment of human disease and the improvement of human biological systems. Previous papers have explored theoretical designs or scaling studies for medical nanorobots including artificial mechanical red cells (respirocytes [41]), artificial mechanical white cells (microbivores [42]), artificial mechanical platelets (clottocytes [43]), nanorobotic pharmaceutical delivery devices (pharmacytes [44]), dental nanorobots (dentifrobots [45]), and an artificial nanomechanical vascular system (vasculoid [46]). This paper presents the first technical scaling study for a true cell repair nanorobot. Called chromallocytes,* these still-hypothetical mechanical nanorobots would be infused into the human body, travel to a cell, enter the cell nucleus, remove the existing set of chromosomes and replace it with a new set, then exit the body, a process called “chromosome replacement therapy” or CRT. As perhaps the ideal gene delivery vector, chromallocytes could provide a complete and permanent cure for almost all genetic diseases by replacing damaged or defective chromosomes in individual living cells with a new set of artificially manufactured chromosomes that are defect-free copies of the originals. Cell targeting would be virtually 100% efficient and complete. Full removal of the original DNA avoids any possibility of iatrogenic aneuploidy (possessing an abnormal number of chromosomes in the nucleus) which is a leading cause of spontaneous miscarriages [47], genetic diseases such as XYY syndrome [48] and congenital heart disease [49], and is a hallmark of many human cancer cells [50]. ---------------------------------------------------------------------------------------------- * Chromallocytes (pronounced “crow-MAL-oh-sites”) are nanorobots capable of chromosome exchange operations inside the living human cell nucleus. The etymology derives entirely from Greek roots. The prefix chroma- (as in chromosome or chromatin, the genetic material present in the nucleus of a cell that is a deoxyribonucleic acid attached to a protein structure base) was taken directly from the Greek word chroma, meaning literally “color,” referring to the fact that the chromosomal components of cells would preferentially stain in early cell biology experiments. The root form -allo- derives from numerous sources, including the Greek roots allage (“change”), allasso or allassein (“to change,” “to exchange”), allos (“other”, “another”, or “changed”), allothi (“elsewhere”), allotrios (“another’s”), and allelon (“of one another”). The suffix -cyte derives from the Greek -kytos (noun: “a hollow”) or -cyto, a combining form meaning “of a cell” or “cells”. Hence “chromallocyte” literally means “a chromosome-exchanging cell”. ---------------------------------------------------------------------------------------------- After an introductory overview of the human cell nucleus, including relevant physical aspects of DNA and chromosomes, the basic chromallocyte scaling design is presented, followed by an exemplar mission description and a brief analysis of special situations and mission design issues involving nanorobotic chromallocytes. The proposed design is complex and likely to be modified, at least in part, as further details of human biology are discovered. As a scaling study, this paper serves mainly to demonstrate that all systems required for mechanical chromosome exchange operations could fit into the stated volumes and could apply the necessary forces, deploy the needed chemical substances, and perform all essential functions within the given power, space and time allotments. This scaling study is neither a complete engineering design nor a formal design proposal for a future nanomedical product. Rather, the purpose here is merely to examine a set of appropriate design constraints, scaling issues, and reference designs to investigate whether or not the basic idea of a chromosome replacement device might be feasible, and to determine key limitations of such machines, as an exercise in theoretical applied science [51e]. Issues in nanorobot biocompatibility, including immune system evasion, have been extensively discussed elsewhere [40-42]. The reader should note that utilization of this nanomedical device as described will require a vast infrastructure of mature medical nanotechnology that does not yet exist. The development of such an infrastructure will proceed in parallel with ongoing efforts to design and build nanofactories [52] capable of fabricating and assembling medical nanorobots [53]. The existence of chromallocytes, some decades hence, thus implies the existence of the necessary infrastructure that is enabled by the same molecular manufacturing technology. 2. Basic Structure of the Cell Nucleus
The cell nucleus, 5-8 microns in diameter for a 20 micron tissue cell and up to 10 microns for a fibroblast, is the largest cellular organelle. It is the only organelle that is voluminous enough, in theory, to admit a micron-scale medical nanorobot into its interior. The nucleus is usually a large spherical or ovoid structure consisting of nucleoplasm surrounded by its own nuclear membrane within the cytoplasm of the cell, although its shape generally conforms to the shape of the cell. For example, if a cell is elongated, the nucleus may be extended as well [54]. Almost all cells contain a single nucleus, whose primary function is the storage and expression of genetic information. However, a few cell types have multiple nuclei of similar size, such as skeletal muscle cells, osteoclasts, megakaryocytes, and some hepatocytes [55]. A few cell types have no nucleus, such as red blood cells, platelets, keratinized squamous epidermal cells, and lens fibers.
2.1 Nuclear Envelope
The nuclear envelope enclosing the nucleus is a lipid bilayer similar in composition to that of the cell membrane, except that it is a double-layered membrane which is topologically more convenient for dissolution during mitosis and subsequent reassembly from vesicles. The nuclear envelope disassembles at the onset of mitosis and is reassembled at the end of mitosis [56]. Each of the two lipid bilayer membranes is 7-8 nm thick. The outer nuclear membrane (ONM) is occasionally continuous with the rough endoplasmic reticulum (ER) and is almost entirely surrounded by it. Like the rough ER, the ONM is often studded on its outer surface with ribosomes involved in protein synthesis [57]. Intermediate filaments extend outward from the ONM into the surrounding cytoplasm of the cell, anchored on the other end to the plasma membrane of the cell or to other organelles, thus positioning the nucleus firmly within the cell and increasing its mechanical stiffness almost tenfold [58].
The perinuclear space (or perinuclear cisterna) between the two lipid membranes ranges in width from 10-70 nm but is usually a gap of 20-40 nm. This fluid-filled compartment is continuous with the cisternae of the rough ER, thus providing one possible avenue for transporting substances between the nucleus and different parts of the cytoplasmic compartment.
Another distinctive feature of the nuclear envelope is the presence of numerous nuclear pores, small cylindrical channels with eightfold symmetry that extend through both membranes and provide direct contact between cytoplasm and nucleoplasm [59-62]. Each pore complex marks a point of fusion between the inner and outer membranes. Elements of the cytoskeleton external to the nucleus appear to be attached to many pores, possibly allowing direct mechanical regulation of pore activity [63, 64]. Each nuclear pore complex is a huge multimolecular assemblage measuring 70-90 nm in diameter, with a mass of 125 million daltons, ~34 times the size of a ribosome. Up to 100 different nucleoporin protein molecules make up the structure [65]. Early experiments with passive gold particles showed that cytoplasmic particles with diameters of 5-6 nm passed through the pores into the nucleus in ~200 sec, those with diameters of 9-10 nm took ~104 sec, but particles >15 nm were excluded [57]. Closer examination has revealed that the pores are actually large enough to allow the passage of substrates as large as 23-26 nm [59, 65], but this is still much too narrow for nanorobots or their flexible robotic protuberances to pass through without damaging the mechanism. The nuclear localization sequence (NLS), a molecular tag consisting of 1-2 short sequences of amino acids, marks cytoplasmic proteins for active transport through the nuclear pores. Small (~40 nm) arm-like import receptors (cytoplasmic filaments) ringing the mouth of the pore bind to a protein cargo tagged with an NLS, then flex toward the pore to shove the cargo into the opening [66-68]. The density of pores across the surface of the nuclear envelope varies greatly, depending mainly on cell type and the amount of RNA being exported to the cytoplasm. Values range from 3-4 pores/micron2 in some white cells up to 50 pores/micron2 in oocytes with a theoretical maximum density of 60 pores/micron2 [57]. A typical ~20 micron human cell has 2000-4000 pores embedded in its nuclear surface [65], a mean density of 10-20 pores/micron2. Pore structures may protrude at most ~100 nm into the nucleoplasmic space. The nuclear cortex is an electron-dense layer of intermediate filaments (composed of the nuclear lamins common to most cell types) on the nucleoplasmic side of the inner nuclear membrane (INM) [65]. The cortex, also called the nuclear lamina or karyoskeleton, is up to 30-40 nm thick in some cells but is difficult to detect in others [57]. Its proteinaceous fibers are arranged in whorls that may serve to funnel materials to the nuclear pores for export to the cytoplasm. These fibers may also be involved in pore formation. The nuclear cortex helps to determine nuclear shape, and also binds to specific sites on chromatin [69] (the form taken by chromosomes between cell divisions), thereby guiding the interactions of chromatin with the nuclear envelope [70]. Chromatin binding sites on the nuclear cortex avoid the immediate vicinity of nuclear pores to ensure unobstructed passage of materials through the pores [70]. 2.2 Nuclear Interior, Chromosomes and DNA
The nucleoplasm is the semifluid matrix in the interior of the nucleus. It contains some condensed but mostly extended chromatin as well as a dynamic structural nuclear matrix [71] of nonchromatin (mostly protein) material; 398 distinct nuclear matrix-associated proteins comprising and attached to the matrix had been catalogued as of 2005 [72], many of them cell-specific [72, 73]. Chromosomes assume a highly condensed (compact) state as the cell prepares to divide, but after mitosis most of the chromosomes relax into a highly extended state that pervades most of the nucleoplasm. During interphase (e.g., between cell divisions), individual chromosomes occupy discrete territories [74-76] within the nucleus that may range up to 3-5 microns in diameter, organized in a radial distribution with the most gene-dense chromosomes located toward the center of the nucleus [77]. The structure and location of these territories varies by cell type and mitotic stage [78, 79], and may be arranged in the same spatial order as is found in the wheel-shaped ring aggregate known as the chromosome rosette at the time of mitotic prometaphase [80]. Note, however, that these territories are not rigid. Changes in the relative positions of chromosomal territories often occur at speeds of 0.3-0.4 nm/sec, and intraterritorial movement and flexing of subchromosomal foci measuring 400-800 nm in diameter have also been observed [81, 82]. Multiple compact chromatin domains within each territory are surrounded by interchromatin space that is largely devoid of DNA [83, 84]. The nucleosol, or fluid component of the nucleoplasm, contains salts, nutrients, and other needed biochemicals, and a number of different granules are also present [85]. Two unbranched polymeric chains of deoxyribonucleic acid (DNA), with each strand comprised of a linear sequence of nucleotides on its own sugar-phosphate backbone and joined to the other strand via hydrogen bonds between complementary nucleotide bases on opposing strands, constitutes a single molecule of duplex DNA, aka. double-stranded DNA or “dsDNA”. (A nucleotide has three parts: (1) a nitrogen-containing pyrimidine or purine base (A, C, G, T), (2) a five-carbon deoxyribose sugar, and (3) a phosphate group that acts as a bridge between adjacent deoxyribose sugars.) Besides the hydrogen bonding between base pairs (bp), dsDNA is also stabilized by van der Waals forces and by hydrophobic interactions between the nitrogenous bases and the surrounding sheath of water. Each very long molecule of dsDNA, forming the familiar ~2.3-nm-diameter [86] double helix, constitutes a single haploid genome whose length is measured in base pairs (pairs of complementary nucleotide bases, one on each strand of the duplex). The second column of Table 1 lists the number of base pairs per copy of each haploid chromosome found in the human nucleus. There are two copies of each haploid chromosome in a diploid chromosome pair, and there are 23 diploid pairs in a human genome, so each nucleus in a human cell contains 46 haploid chromosomes or 23 diploid chromosomes with a total duplex-DNA contour length of ~2 meters (at 0.335 nm/bp [87, 88]). The DNA contains the genes of the cell, and all 25,000-30,000 human genes [89, 90] are represented, though not expressed, in each nucleated somatic cell. Each chromosome in a human nucleus includes a mass of protein roughly equaling the mass of the DNA. There is very little, if any, free DNA in the nucleus. Chromatin is the complex of DNA and protein in the nucleus of the non-dividing (interphase, non-mitotic) cell. The chromatin takes two forms: Euchromatin, which is dispersed and loose, occupying most of the nucleus, and in which genes are being expressed; and heterochromatin, which is densely packed or “condensed”, and in which genes are not being expressed. (Fully condensed mitotic chromosomes are transcriptionally inert, as cells virtually cease transcription during mitosis.) Both forms are present in living cells during interphase. In its most relaxed state, euchromatin resembles a network of bumpy threads weaving their way through the nucleoplasm.
The proteins associated with chromosomes are of two types: Histones and nonhistones. Histones. Chromatin is composed of roughly equal amounts of negatively charged DNA and globular histone proteins (basic proteins that carry a positive charge at the normal pH found in the cell [85]). DNA is bound to the histones through electrostatic forces between the negatively charged phosphate groups in the DNA backbone and positively charged amino acids (e.g., lysine and arginine) in the histone proteins. Five classes of histones were originally characterized, based on their relative proportions of lysine and arginine. H3 and H4 are the most conserved proteins in all of evolution. H2A and H2B have some species-specific differences. These four histone types, called the core histones, are small proteins, typically 11-15 kD. Then there is H1, actually a set of several rather closely related proteins with overlapping amino acid sequences. The H1 “linker” histones show appreciable variation between species and even between tissues, and apparently are entirely absent from yeast. Among histones, H1 is the largest, about 25 kD [87]. Histone proteins are sometimes modified by the addition of acetyl, methyl, or phosphate groups, altering the strength of the bonding between the histones and DNA. Such modifications are usually associated with the regulation of biological processes such as DNA replication, gene expression, chromatin assembly and condensation, and cell division [88]. The core histones are organized into ellipsoidally-shaped histone octamers. In human cells a short ~200 bp segment of dsDNA (~67 nm contour length) is coiled around the curved surface of each octamer (like a solenoid winding), completing about 1.8 complete turns. Each histone octamer is composed of two copies each of H2A, H2B, H3, and H4, the core histones. Stoichiometrically, the core histones are present in equimolar amounts, with 1 molecule of each per ~100 bp of DNA. H1 is present in about half the amount of a core histone (i.e., 0.5 molecule per ~100 bp of DNA) and lies external to the particle, since all of the H1 can be removed from chromatin without affecting the structure of the particles. The DNA+histone particles, called nucleosomes, are the fundamental units of chromatin, connected like beads on a string by a DNA molecule that winds around each of them. Approximately 166 base pairs are bound to the nucleosome (~146 tightly bound to the core particle and the remaining 20 associated with the H1 histone), while the DNA between two nucleosomes is called the linker segment and includes the rest of the ~200 bp. The average cell nucleus contains 25 million nucleosomes, each ~6 nm tall and ~11 nm in diameter. H1 is dynamically associated with chromatin, with each H1 molecule binding chromatin for ~1 minute, then falling off and freely diffusing through the nucleoplasm until it encounters another binding site [91]. The core histones typically reside on chromatin for several hours [92]. The predicted mass of the nucleosome is 262 kD, with a protein/DNA mass ratio of ~1. This mass includes ~200 bp of DNA of mass 130 kD, a histone octamer consisting of two H2A at 28 kD, two H2B at 28 kD, two H3 at 30 kD, and two H4 at 22 kD (giving a 108 kD octamer), plus a single H1 at 24 kD. The experimentally measured mass is usually in the range 250-300 kD, with a protein/DNA ratio of up to ~1.2; the additional variable protein amount represents small amounts of nonhistone proteins (see below) associated with the nucleosomes. Nonhistones. The nonhistones are all the other proteins of the chromatin, presumed more variable between species and tissues, and they comprise a relatively smaller proportion of the mass than the histones. They also comprise a much larger number of proteins, so that any individual protein is present in amounts much smaller than for any histone. The nonhistones perform functions concerned with gene expression and with higher-order structure. RNA polymerase may be considered a prominent nonhistone. The high-mobility group (HMG) proteins are a discrete and well-defined subclass of nonhistones, at least some of which are transcription factors. HMG proteins also exhibit stop-and-go binding to chromatin, but with a residence time of only seconds and with a significantly larger unbound fraction [93]. Since some nonhistone proteins are more durably bound to the chromatin, many of these too will be extracted and replaced in the nucleus by the chromallocyte during the chromosome replacement operation. If each nucleosome occupies a cylindrical volume of ~570 nm3 per 200 bp, and if associated nonhistone proteins increase this volume by up to an additional 20%, then this yields an estimated 684 nm3 of histone+nonhistone protein volume per 200 bp of DNA. Applying this estimate to the base pairs present in each human chromosome yields the per-chromosome and total volume estimates listed in the third column of Table 1. Note that while nucleotide bases in living cells are sometimes modified by the addition or alteration of chemical groups (most commonly, by the methylation of the 5’ carbon atom of cytosine), only 2-8% of the 5’ cytosine carbon atoms are typically methylated in vertebrates – an amount of methylation which in human DNA would change the average base pair mass by less than 0.07% [88]. The fact that individual chromosomes in decondensed euchromatin cannot be readily distinguished in the nucleus except by direct chemical inspection underlies the logic of chromosome replacement therapy (CRT). Rather than attempting to exchange a single chromosome among the 23 diploid pairs or to repair a specific base pair sequence on a specific chromosome strand (Section 6.8), CRT does not attempt to sort through or chemically scan individual chromosomes in situ, but simply exchanges them all at the same time in a single replacement operation. The displacement volume of old chromosomal material to be removed from the cell nucleus by the chromallocyte is taken as 20.654 micron3 for the slightly larger chromatin load that is present in the typical cell nucleus of a human female (Table 1). Additionally, the mass of the RNA present in the eukaryotic cell nucleus is taken as ~10% of the mass of the DNA – there is ~10 times more RNA than DNA in the human cell [39zz], but most of this RNA is present in extranuclear ribosomes (incorporating rRNA) and tRNA with only ~3% of cellular eukaryotic RNA present as mRNA [87], of which we assume perhaps one-third is intranuclear, given the typical >30:1 cytoplasm/nucleoplasm ratio. Much of the intranuclear RNA consists of nascent chains still associated with the template DNA. Conservatively adding the entire nuclear RNA volume of ~28.5 nm3 per 200 bp (~0.86 micron3 for the cell nucleus of a human female) brings the total displacement volume of material to be removed to ~21.5 micron3, which includes 9.46 micron3 of DNA/RNA and 12.04 micron3 of protein. The empirical buoyant density of DNA is ρDNA = 1.660 + 0.00098(GC%) gm/cm3, where GC% is the fractional GC nucleotide content expressed as a percentage [87]; GC% ~ 42% for mammalian DNA [87], hence ρDNA ~ 1.70 gm/cm3. Chromatin buoyant density is ~1.40 gm/cm3 [94], so the density of chromosome-related protein is ~1.10 gm/cm3, hence the total ~2.93 x 10-11 gm of chromosome-related mass to be removed from the typical human cell nucleus includes ~1.61 x 10-11 gm of DNA/RNA and ~1.32 x 10-11 gm of protein. If the displacement volume of new chromosomal material to be delivered and implanted into the cell nucleus by the chromallocyte is conservatively approximated as the same ~21.5 micron3, then an estimate of the required nanorobot onboard storage capacity requires knowledge of the maximum chromatin packing density that can be achieved. Chromatin in living cells is most compact during mitotic metaphase – metaphase chromosomal density is typically 0.043 gm/cm3 in plants and animals [95] and 0.081 gm/cm3 in humans [96, 97], giving packing densities relative to chromatin buoyant density of only 3.1% and 5.8%, respectively. Similarly, dividing total chromatin volume of 21.5 micron3 by the average volume of the human nucleus [39a] of 268 micron3 gives an 8.0% packing density for in vivo euchromatin. However, a density of 0.530 gm/cm3, a natural 38% packing density, has been reported in pre-extrusion hen erythrocytes [98]. Duplex DNA packed into the viral capsids of bacteriophages occupies about twice the excluded volume of the free double helix which gives a packing fraction of ~50% [99], though this figure might not be considered strictly comparable to the others because the viral material lacks the associated eukaryotic proteins. For such proteins, we note that natural protein-interior packing densities are typically ~60%-85% [100] and the volumetric packing factor of closely-packed spheres of equal radius is (3π2/64)1/2 ~ 68% [101]. For convenience in this scaling study, we assume that a comparable chromatin packing density of fpacking = 54% (49%, if mRNA is excluded) can be achieved while preserving chromosome integrity and while leaving sufficient protein-associated water to avoid denaturation (possibly including small amounts of extractable stabilizers [102]). This gives a required chromallocyte onboard storage volume of 40 micron3 to transport a single complete set of human female chromosomes including all genomic DNA and appurtenant proteins. Note that the volume of chromosome-related material in the nucleus which is to be exchanged may vary slightly from cell to cell depending upon various factors such as cytotype, cell cycle status, local protein synthesis activity level, and so forth.
2.3 Nucleolus
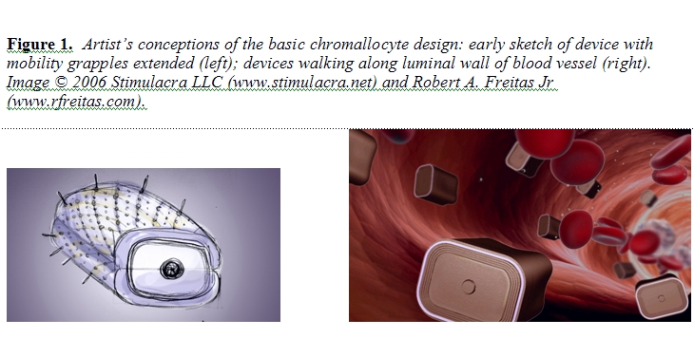
Several subnuclear organelles are known [103]. The largest and most prominent is the nucleolus, a highly coiled structure associated with numerous particles but not surrounded by a membrane [104]. The nucleolus is a ribosome-manufacturing machine: assembly of precursor ribosomal subunits within the nucleolus requires ~1800 sec, while the complete assembly of a large ribosomal subunit (needing only protein to make a completed ribosome) takes ~3600 sec [70]. The nucleolus is composed of DNA, RNA, and proteins. It also has a granular component (each granule ~150 nm thick) and a fibrillar component, and a variable internal structure [104]. The granular component consists of ~15-nm particles that are ribosomal subunits in the process of maturation. The fibrillar component consists of rRNA molecules that have already become associated with proteins to form fibrils with a thickness of ~5 nm. The size of the nucleolus correlates with its level of activity. In cells characterized by a high rate of protein synthesis and hence by the need for many ribosomes, the nucleolus can occupy 20-25% of nuclear volume (3-5 micron diameter in a 20-micron cell), mostly comprised of the granular component. In less active cells, the nucleolus is much smaller – as small as 0.5 micron in a mature lymphocyte [104]. Nucleoli are frequently located at or near the nuclear envelope, adhering directly to the nuclear lamina or attaching to it by a pedicle. In nuclei having a centrally located nucleolus, the nuclear envelope is folded to form a nucleolar canal that is in direct contact with the nucleolus [104]. Most human nuclei contain only one nucleolus, except for hepatocyte nuclei which may contain more than one nucleolus [55]. The number of nucleoli in a eukaryotic cell nucleus normally is determined by the number of chromosomes with secondary constrictions, or nucleolus organizer regions (NORs). The human genome contains five NORs per haploid chromosome set, or 10 NORs per diploid nucleus, each located near the tip of a chromosome. However, instead of 10 separate nucleoli, the typical human nucleus contains a single large nucleolus representing the association of loops of chromatin from the 10 separate chromosomes with NORs. The DNA from the remaining diploid chromosomes is distributed in specific regions throughout the nucleoplasm. During mitosis, the chromosomes condense into a more compact form and the nucleolus shrinks, then disappears altogether. A cell undergoing mitosis thus has no nucleolus and synthesizes no rRNA. Once mitosis is complete, the nucleolus reappears. During CRT, appropriate molecules are released into the nuclear interior by the chromallocyte to compel the deconstruction of the nucleolus without triggering mitosis or apoptosis (Section 5.2). 3. Chromallocyte Structure and Function This Section describes the basic structure of the chromallocyte including all important subsystems. For computational convenience in performing this scaling study, the constitutive equations assume only circular or rectangular component geometries. The anticipated nonangular and noncircular surfaces in the actual nanorobotic device (e.g., curved hulls, rounded corners, etc.) will cause minor deviations from the calculated sizes and volumes reported here but should not significantly affect the overall design. 3.1 Overall Nanorobot Structure The chromallocyte (Figure 1) is a lozenge-shaped motile cell-repair nanorobot having an estimated external (displacement) volume of 69.250 micron3, a minimum (non-distended) surface area of 102.778 micron2, an unloaded “dry” mass of 80.239 pg (incorporating ~4.0 x 1012 atoms of nanomachine structure, mostly diamondoid) and a fully-loaded mass (including “wet” cargo) of ~109.5 pg. The nanorobot measures 4.18 microns wide, 3.28 microns tall, and 5.05 microns in length, with a maximum transdevice diameter (along a rectangular prismatic diagonal) of 7.33 microns. These values are the result of a simultaneous partial optimization of several important design constraints as described below.
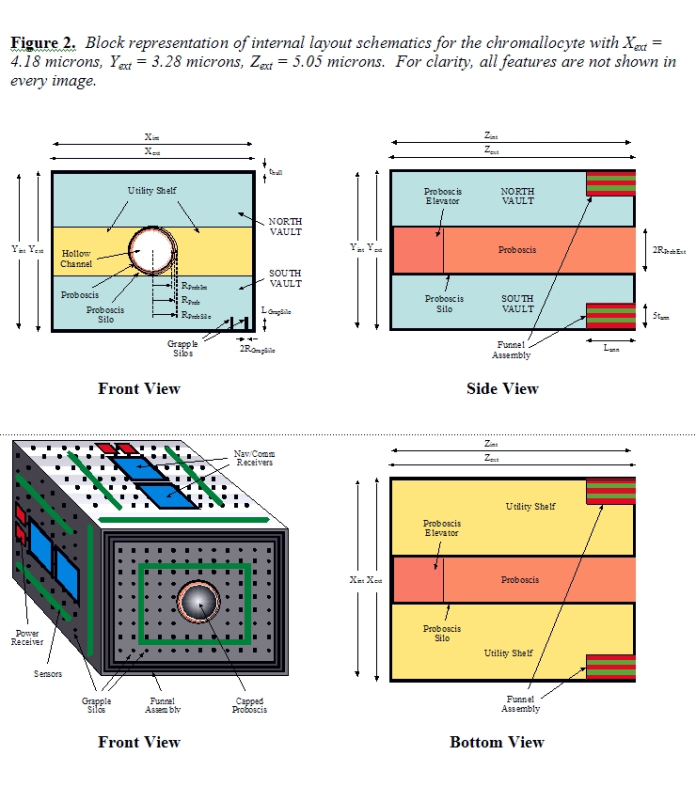
The transdevice diameter for free-floating bloodstream nanorobots [39b] is normally limited to ~4 microns to avoid trapping in the smallest-diameter human capillary vessels [39c, 40a]. Chromallocytes are not allowed to free-flow and are restricted to vascular surfaces when traversing the bloodstream, both during infusion and extraction from the body at the end of the mission (Section 5.2). At 69.250 micron3 they remain smaller in volume than either erythrocytes (~95 micron3 red cells) or granulocytes (~1000 micron3 white cells) which regularly traverse the microvasculature. Cell intrusiveness [40c] is acceptable since these nanorobots are less than 1% of typical tissue cell volume, though up to ~25% of nucleus volume. Figure 2 shows the general structure of the chromallocyte nanorobot. Taking the device’s lozenge shape as a rectangular prism of external dimensions Xext, Yext, and Zext with hull wall thickness thull, then the interior dimensions are Xint = Xext – 2thull, Yint = Yext – 2thull, and Zint = Zext – 2thull. This gives an external volume of Vext = XextYextZext, an internal volume of Vint = XintYintZint, and an external surface area of Sext = 2(XextYext + XextZext + YextZext).
3.2 Proboscis Manipulator A single large axially-positioned manipulator called the Proboscis, of length LProb and outer cylindrical radius RProb, is used to collect old chromatin from the nucleus and later to transfer new chromatin from the nanorobot internal storage into the cell nucleus by conduit flow. When stowed and not in use, the Proboscis resides in a large silo of length LProbSilo and outer cylindrical radius RProbSilo, with a silo wall thickness tProbSilo = RProbSilo – RProb and with LProbSilo = 1.25 LProb to accommodate elevator motors, gearing, power connections, and so forth needed to control and drive Proboscis motions. Assuming the available mechanical energy density of this support machinery [39d] is dpower ~ 109 W/m3 and taking LProb = 4 microns and RProb = 0.55 micron, then the maximum mechanical energy nominally available to drive Proboscis motions is EProbMech ~ (0.25 LProb) (πRProb2) dpower = 950 pW which can be provided by onboard power systems (Section 3.6). The Proboscis is a manipulator similar to the class of telescoping manipulator [39e] originally described by Drexler [51d], except that the Proboscis contains no telescoping joints. Instead, it is constructed solely of pairs of canted rotating joints arranged such that their relative rotation produces a change in angle of the manipulator. Some length change can be provided by the rotating joints, but Proboscis extension beyond the nanorobot hull perimeter is controlled primarily by the elevator mechanism in the silo. The Proboscis has relative dimensions similar to those of the grapple shown in Figure 4 and is also driven by the forced rapid rotation of internal drive shafts. But the Proboscis has a smooth cylindrical exterior surface and a hollow interior conduit of radius RProbInt = 0.50 micron to accommodate an outflowing semiliquid chromatin payload, and terminates in an irising valve that may be opened to allow outflow or closed to prevent entry of environmental fluids into the nanorobot interior. During initial use, the Proboscis is extended outward from the slightly convex prow of the nanorobot into the nuclear interior. Presentation semaphores [39f] on the external surface of the manipulator are rotated to their chromophilic (chromosome-binding) position, producing a large adhesioregulatory surface [40b] to which chromatin will strongly adhere (see below). Since the typical tissue cell nucleus is ~5 microns in diameter and the front end of the nanorobot is slightly inserted through the nuclear envelope into the nucleoplasm, a Proboscis of length LProb = 4 microns provides an essentially complete trans-nuclear reach in nuclei of normal size (see Section 6.5). Penetration of the opposite wall of the envelope is avoided by employing an apical-mounted sensor that detects the presence of a resistive lamin-mesh membrane, halting further extension of the manipulator. Additionally, smaller adhesioregulated chromophilic tines may be laterally extruded from helicoid silos in the exterior Proboscis wall producing a muricate surface resembling a sticky bottlebrush. This enlarges the effective chromophilic volume that is attached to the manipulator and thus increases the number of potential binding points between Proboscis and chromatin strands, reducing spool time. The spiny Proboscis is then slowly rotated and laterally gyrated, spooling any detached chromatin present inside the nucleus into an ellipsoidal bolus wrapped around the manipulator. The bolus is subsequently withdrawn from the cell by envelopment within a telescoping funnel assembly (Section 3.3) that is sealed, then retracted, forcing the old chromatin into vacated vault volumes as the new chromatin flows out into the nucleus through the Proboscis interior conduit. A crude estimate of the power requirement for Proboscis rotation during spooling of the chromatin bolus is provided by the Stokes drag power P = 6πηRv2 for spheres in fluid, which generally gives higher values than the more complex Lighthill drag power formula for cylinders translating in fluid, for this geometry [39h]. At the inception of spooling, a Proboscis of radius R = RProb = 0.55 micron is conservatively assumed to be inserted into a packed-chromatin-like fluid of absolute viscosity ηchromatin = 5 x 107 kg/m-sec (Section 3.4) and rotated at a frequency of υ Hz with a tangential velocity of v = 2πRυ, giving a starting drag power of Pstart ~ (6200 υ2) pW. Near the end of spooling, the Proboscis plus an attached bolus (presumed spherical) of radius R ~ wbolus/2 = 1.61 micron (Section 3.3) is assumed to be rotating in a cytoplasm-like fluid of absolute viscosity ηcytoplasm = 100 kg/m-sec (Section 3.3), giving an ending drag power of Pend ~ (0.31 υ2) pW. A typical spooling profile might initiate Proboscis rotation at υ ~ 0.1 Hz consuming Pstart ~ 62 pW to begin the spooling, slowly accelerating to υ ~ 1 Hz consuming Pend ~ 0.3 pW by the completion of spooling. The initial 0.1 Hz rotation rate of the Proboscis represents a tangential velocity of ~0.3 micron/sec, comparable to peak chromosome transport speeds during mitosis of ~0.1 micron/sec requiring ~0.1 pN of force [105]. Note that viscous force dominates inertial: ~0.1 pJ accelerates a 100 pg mass to 1 micron/sec in 1 second. Total spool time is scaled by the length of the longest chromosome to be spooled since all strands are wound simultaneously. Assuming 20 equally spaced binding points to chromosome #1 (length 82.1 mm, Section 2.2) or ~500 binding points to all strands and assuming 100% euchromatin, and noting that chromatin looped between sequential binding points is doubled over when spooled, a maximum length of 2.05 mm must be spooled. Taking a mean bolus diameter of 2.16 micron during spooling and a mean spooling rotation rate of ~0.3 Hz, spooling requires ~300 turns and an estimated ~1000 sec to complete. Chromophilic presentation semaphores will require the design [39dd], simulation [106], and fabrication of general-purpose reversible binding sites for chromatin. Such artificial binding sites may be similar to the many known biological receptors and binding sites on proteins having similar function. These are of two types. First, there are the sequence-specific DNA binding sites for base-paired nucleotides as found in restriction endonucleases [107] and in gene regulatory proteins such as the DNA-binding domain of the yeast transcriptional activator GAL4 [108]. Second, there are sites enabling non-specific DNA binding by endonucleases [107] which permit one-dimensional diffusion of proteins along DNA [109]. Conformational changes in these binding proteins commonly result in the expulsion of solvent molecules from the interface to allow for more intimate contacts, both during specific [110] and non-specific [111] binding to undamaged DNA. Nonspecific DNA binding by other proteins [112] and peptides [113] is well-known, including, for example, the single-stranded DNA-binding protein (SSB) from E. coli for undamaged ssDNA [114], the binding site for ssDNA and dsDNA in the highly-conserved RecQ helicase protein [115], zinc-finger DNA-binding domains [116], the chromodomains of CHD proteins [117], the DNA binding subunit (Ku) of DNA-dependent protein kinase (DNA-PK) [118] that binds to DNA double-strand breaks [119], and the 122 amino acid region in human XPA (xeroderma pigmentosum group A) protein [120] that recognizes damaged single-strand DNA as part of the nucleotide excision repair system [121]. Binding sites for phosphate analogous to those found in DNA-cleaving phosphodiesterase enzymes [122], phosphate-binding adenovirus type 5 E1A protein [123], the phosphate binding site of 2-deoxyribose-5-phosphate aldolase (DERA) of E. coli [124], or in the p53-related AML1-CBF beta complex that clamps the phosphate backbone between the DNA major and minor grooves [125], could allow direct chromophilic binding to the DNA deoxyribose-phosphate backbone. Other nonspecific binding surfaces for DNA are found on histones [126], inorganic crystals [127], and elsewhere [128]. Binding sites for histones include HCV-polyprotein-(1343-1379) for core histones [129], murine antiidiotope BII 2.1 monoclonal antibody for core histone H3 [130], the double-chromodomain recognition site for the methylated histone H3 tail [117], and NASP (nuclear autoantigenic sperm protein [131] and nucleoplasmin [132] binding sites for link histone H1. Binding sites analogous to those for nuclear matrix proteins such as nuclear mitotic apparatus protein (NuMA) which might be present in protein p73 (allowing it to preferentially bind to NuMA) [133] could provide supplemental though indirect chromatin adhesivity for designed chromophilic presentation semaphores.
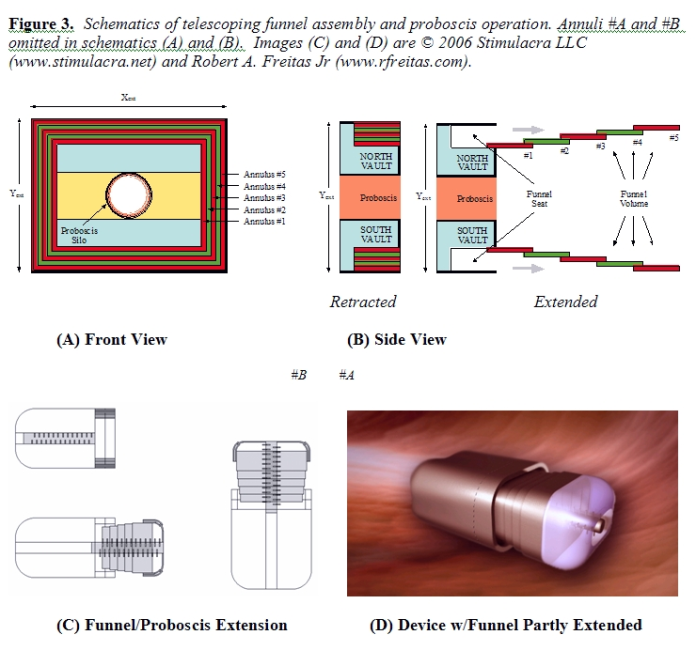
3.3 Funnel Assembly After the Proboscis has spooled the nuclear chromatin into an ellipsoidal bolus, the funnel assembly is extended out into the nucleoplasm, surrounding and ultimately fully enclosing the bolus. As illustrated in Figure 3A/B, the core funnel assembly is composed of Nann = 5 nested annuli. Each nested annulus (#1 through #5) has length Lann = 1 micron, wall thickness tann = 35 nm, and a maximum extensibility of fext = 80% (i.e., adjacent annuli retain 0.2 micron lengthwise overlap at full extension). Drive shafts located internally to each annular plate turn worm gears engaged on adjacent plates, forcing one plate to slide across the other in the desired manner. Cam followers on divided segments of the flexible outermost two annuli #A and #B (Figure 3C) are designed to slide inward to form a circular irising aperture that can establish and maintain full perimeter contact with the outer surface of the Proboscis, creating an interior void large enough to contain the entire chromatin bolus (Figure 3D). These two annuli extend to form a watertight cap across the open end of the funnel. The total exposable surface area of annuli #A and #B assuming 80% extensibility is ~23.9 micron2, exceeding the ~13.7 micron2 cross-sectional area of the fully extended funnel mouth aperture by 74%, hence the flexible irising segments comprising annuli #A and #B can overlap by up to 43% in surface area and still perform their required function. This function includes pressurization of enclosed contents up to 1000 atm to assist waste chromatin pumping. A spherical tank of radius Rtank = Xext/2 = 2.09 microns, wall thickness twall = 35 nm, and very conservative working stress σw ~ 1010 N/m2 (~0.2 times the failure strength of diamond [39p]) has a bursting pressure pburst ~ 2 twall σw / Rtank ~ 3300 atm [39q]. While the 35 nm thick annular walls are not entirely solid, only twall ≥ 11 nm is required to withstand the specified maximum 1000 atm load. To geometrically accommodate both the Proboscis silo and the stowed funnel annuli within the nanorobot structure, we require: Xint ≥ 2RProbSilo + 2(Nanntann) + 2(LGrapSilo – thull) Yint ≥ 2RProbSilo + 2(Nanntann) + 2(LGrapSilo – thull) Zint ≥ LProbSilo – thull The length of the fully-extended funnel assembly LExtFunnel = fextNannLann must exceed the length of the fully extended Proboscis (LProb), and the width of the fully-extended funnel assembly wExtFunnel must exceed the anticipated width of the fully-spooled chromatin bolus wbolus = 2(RProb + tbolus). The bolus thickness is given by tbolus = (Vchromatin/πLbolus) + RProb2)1/2 – RProb, where Vchromatin ~ 21.5 micron3, the length of the spooled bolus is Lbolus ≤ LProb [39r], wExtFunnelX = Xint – 2thull in the X dimension, and wExtFunnelY = Yint – 2thull in the Y dimension; hence we require: Lann ≥ LProb / fextNann wExtFunnelX ≥ wbolus wExtFunnelY ≥ wbolus
In the baseline design with the aforementioned parameters, wExtFunnelX = 3.98 microns and wExtFunnelY = 3.08. For Lbolus = LProb, wbolus = 3.22 microns which slightly exceeds wExtFunnelY; but this may be acceptable because the enclosed funnel volume of Vfunnel = 43.63 micron3 slightly exceeds πRProb2LProb + (Vchromatin / fpacking) = 43.62 micron3. If this funnel volume proves insufficient, more volume can be made available by introducing more structural flexibility into the design and a slight outward curve to each deployed nested annulus. The drag power dissipated by two sliding contacting diamond surfaces with interfacial velocity vplate and contact area Aplate has been estimated [51a] in the case of nanoscale bearings as Pdrag ~ 1100 vplate2 Aplate (watts). Each annulus has a maximum contact area Aplate ~ 2(Xint + Yint) Lann ~ 15 micron2, hence load-free motion at vplate = 40 micron/sec (allowing a 4-micron extension in ~0.1 sec) requires Pdrag = 0.00003 pW for each annulus that is in motion. More significant is the Stokes drag power PStokes = 6πηcytoplasmRvplate2 ~ 6 pW, assuming R ~ 2 microns for the entire funnel with all 5 primary annuli operated simultaneously and cytoplasm viscosity ηcytoplasm ~ 100 kg/m-sec [39i]. During funnel retraction to fill the vaults, an additional 26 pW is required to overcome pipe flow resistance (Section 3.4). A 32 pW load requires a 0.032 micron3 motor to drive the funnel assembly, assuming a nanomachinery power density dpower ~ 109 W/m3), hence 0.32 micron3 of funnel assembly motor mechanisms are required to maintain tenfold redundancy in this design. Fluids and macromolecules intruding into the funnel seat during funnel extension are squeezed out of the space during funnel retraction. Semifluid material trapped during retraction of the final segments (annuli #5/A/B) can escape through ported weepholes leading to the external environment that are located circumferentially near the base of the seat, though the seal created by the closure of annuli #A and #B, once established, might be retained intact for the duration of the mission if desired. An alternative design in which nuclear DNA is first digested by intranuclear release of an engineered synthetic nuclease, then the resultant nucleotide digesta are extracted from the nucleus via molecular sorting rotors, in principle eliminating both proboscis and funnel, is infeasible because (lacking the extended sealed funnel assembly or equivalent inflated volume) there is insufficient onboard storage volume (Section 3.4) to contain the necessary two (or more) genomic volumes, and is inadvisable because it relies solely upon uncontrolled diffusion processes to guarantee that none of the original nuclear DNA remains intact.
3.4 Chromatin Storage Vaults New chromatin destined for placement in the cell nucleus is carried in one of two onboard chromatin storage vaults which total Vchromatin / fpacking = 40 micron3 in volume. As the new chromatin is discharged into the cell nucleus through the Proboscis, the old chromatin held in the sealed telescoping funnel assembly is forced into the vacated storage vaults as they are emptied. Two vaults, labeled North and South, are shown in Figure 2. Due to the finite volume of each chromosome and the nondivisibility of new chromosomes destined to be installed in the cell, one might suspect that the 40 micron3 volume of new chromatin would not be precisely divisible into two equal aliquots of 20 micron3 each. (Old chromosomes slated for disposal need not be maintained intact, hence have no such nondivisibility constraint.) However, the two portions can be extremely close in size. For example, using one portion consisting of the 11 diploid chromosomes 1, 3, 5, 7, 9, 11, 13, 17, 19, 21, and 23 that includes 3,025,486,522 base pairs (50.098%) and a second portion consisting of the 12 chromosomes 2, 4, 6, 8, 10, 12, 14, 15, 16, 18, 20, and 22 that includes 3,013,633,516 base pairs (49.902%) results in two almost identical aliquots having an insignificant ~0.02 micron3 variance from an exact 20/20 micron3 split. Vaults are emptied of their contents by backflushing with pumped-in water, forcing the new chromatin out of the vault, or are filled with waste chromatin by pumping water out of the vault, providing a vacuum suction which (with the help of the slowly retracting but sealed funnel assembly) forces the old chromatin into the vault. It may also be necessary to employ a water-impermeable diaphragm or piston mechanism separating new and old chromatin volumes, in order to: (1) avoid water leakage around the chromatin, (2) provide additional ejection force and ensure smooth passage of new chromatin, (3) prevent mixing of old and new chromatin, and (4) serve as a diffusion barrier if nuclease is used to fragment the old chromatin trapped in the sealed funnel (see below). The average flow distance for vault emptying is Lflow ~ (LProb + LVault)/2 ~ 4.5 microns, where LVault ~ 5 microns is the length of a vault of volume VVault = 20 micron3. If the effective flow radius of a vault is Rflow ~ (VVault / πLflow)1/2 ~ 1.13 microns, the Poiseuille forcing pressure is Dp ~1000 atm, and the absolute viscosity of chromatin is ηchromatin = 5 x 107 kg/m-sec (as estimated from typical bacteriophage DNA discharge parameters in which 2.6 x 10-23 m3 of DNA passes through a channel of radius 21 nm and length 54 nm in 200 sec under 50 atm pressure [134]), then the flow time to empty each vault is τflow ~ 8 ηchromatin Lflow VVault / π Δp Rflow4 = 1950 sec and the power draw is Pflow ~ π (Δp)2 Rflow4 / 8 ηchromatin Lflow ~ 26 pW during the 2τflow ~ 3900 sec that is required to empty both vaults at a net flow rate of ΔVflow = 1.03 x 10-2 micron3/sec. An additional πRProbInt2Lflow / ΔVflow ~ 340 sec is required to clear the Proboscis internal channel of the final contents, giving a minimum time requirement of 4340 sec to discharge the new chromatin into the cell nucleus through the Proboscis. Both τflow and Pflow could be dramatically reduced by allowing vaults to be more rapidly emptied through large gated portholes temporarily opened in the vaults’ forward surface. However, the present design which requires new chromatin to flow through the Proboscis allows the physical placement of new chromosomes in selected intranuclear locations within the work envelope of the Proboscis, along with any appurtenant enzymes, messenger molecules, or other biochemical supplements that may be deemed necessary. To empty the vaults of new chromatin, water from the external environment is pumped into the end of the vault most distal from the discharge point via molecular sorting rotors which can pump small molecules such as water against head pressures of up to 30,000 atm [39s]. Establishing a pumping rate of ΔVflow = 1.03 x 10-2 micron3/sec requires the transport of 3.4 x 108 molecules/sec of water. The exemplar 7 nm x 14 nm x 14 nm sorting rotor transports ~106 molecules/sec, hence a minimum of 340 rotors are needed, or 3400 rotors to maintain a systematic tenfold redundancy in the design, occupying 0.333 micron2 of hull space and 0.0047 micron3 of onboard volume. An approximately equal volume is allocated for control and power cabling to the rotors, and for other support structure. A flow rate of ΔVflow for ηwater ~ 10-3 kg/m-sec water at 310 K at a driving pressure of Δp = 1000 atm could be accommodated by an Lpipe = 4 micron pipe having a minimum radius Rpipe = (8 ηwater Lpipe ΔVflow / p Δp)1/4 > 1 nm and a negligible total volume displacement of p Rpipe2 Lpipe ~ 10-5 micron3. (A practical system would use a much larger pipe radius due to the nanoscale stickiness of water [135].) Note that vaults may be packed (at the ex vivo chromosome-manufacturing nanofactory; Section 4.3) with new chromatin organized in layers, with inert immiscible fluid (or alternative mechanical means) separating each layer to discourage commingling and individual whole chromosomes confined to each single packed layer. If vault unloading is pulsed or even alternated between North and South vaults, the order and timing of chromosome discharge can be controlled although the ordinal sequence of discharge from a given vault is established by the loading order during manufacture and cannot be altered in situ. To load the vaults with old waste chromatin, all adhesioregulated tines are retracted and all chromophilic surfaces are switched to chromophobic settings while the distal end (#A/#B) of the sealed funnel assembly is held in close contact with the Proboscis manipulator, maintaining a watertight seal and trapping the waste chromatin inside the funnel volume. The funnel assembly then slowly retracts with enough force to establish up to ~1000 atm compressive pressure (requiring similar or reduced power levels to overcome pipe flow resistance), forcing the waste material into the vaults through large gated portholes temporarily opened in the vaults’ forward surface. Simultaneously, the water that fills the Proboscis internal channel and the vaults is pumped out of the nanorobot to make room for the incoming waste material. Release (then reacquisition) of a small amount of nuclease enzyme into the enclosed funnel volume could detach and significantly fragment the trapped old chromatin, making the waste fluid less viscous and thus faster to pump using less energy, and also eliminating any small residuum of chromatin strands that might remain attached to the Proboscis exterior, funnel interior, or forward hull surfaces.
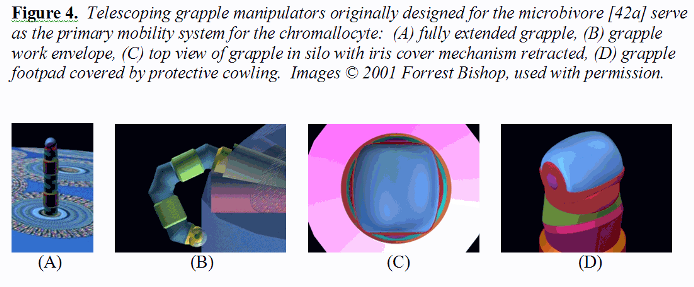
3.5 Mobility System The primary mobility system for the chromallocyte is the telescoping grapple mechanism previously described for the microbivore [42a]. Each grapple is mechanically equivalent to the telescoping robotic manipulator arm described elsewhere [39e, 51d] but is ~2.5 times the length. This manipulator when fully extended (Figure 4A) is a cylinder 30 nm in diameter and 250 nm in length with a minimum 150-nm diameter hemispherical work envelope even on the chromallocyte hull plane (Figure 4B), capable of motion up to 1 cm/sec at the tip at a mechanical power cost of ~0.6 pW at moderate load (or ~0.006 pW at 1 mm/sec tip speed), and capable of applying ~1000 pN forces with an elastic deflection of only ~0.1 nm at the tip.
Each telescoping grapple is housed beneath a self-cleaning irising cover mechanism (Figure 4C) that hides a vertical silo measuring 2RGrapSilo = 50 nm of interior diameter (surrounded by 25 nm thick walls) and LGrapSilo = 300 nm in depth, sufficient to accommodate elevator mechanisms needed to raise the grapple to full extension or to lower it into its fully stowed position. At a 1 mm/sec elevator velocity, the transition requires 0.25 millisec at a Stokes drag power cost (operating in human blood plasma) of 0.0008 pW, or 0.08 pW for 100 grapples maximally extended simultaneously [39ww]. The elevator mechanism consists of compressed nitrogen gas pumped into or out of the subgrapple chamber volume from a small high-pressure sealed reservoir, a pneumatic piston providing the requisite extension or retraction force. A grapple-distension force of ~100 pN applied for a distance of 250 nm could be provided by 25 atm gas pressure in a minimum subgrapple chamber volume of ~104 nm3, involving the importation of ~6000 gas molecules into that volume. Removal of these ~6000 gas molecules from an expanded subgrapple chamber volume of ~105 nm3 induces a complementary retraction force which may be further assisted by cables, springs, and related mechanisms. The aperture of the irising silo cover can be controlled to continuously match the width of the protruding grapple, greatly reducing the intrusion of foreign biomolecules into the silo. Note that although the grapple silo housings protrude a short distance into the interior vault volume, chromatin that is being offloaded or onloaded can flow around these small nubbin-like obstacles without damage provided that the vault-facing surfaces of the silos are curved and atomically smooth (or covered, if necessary, with a chromophobic coating). Silo control and power cables are placed in smooth conduits running along the inside surface of the outermost vault walls. Each grapple is terminated with a reversible footpad ~20 nm in diameter (Figure 4D). For traversing lipid-rich cell surfaces, a footpad may consist of up to 100 close-packed lipophilic binding sites targeted to plasma membrane surface lipid molecules (e.g., binding sites to phospholipids generally [136-138], phosphatidylcholine [139], or other lipids [140]), providing a secure 100-1000 pN anchorage between the nanorobot and the cell surface assuming a single-lipid extraction force of 1-10 pN [39ee]. Footpads may also incorporate binding sites for proteins or glycocalyx carbohydrates as needed. For example, traversing vascular surface or ECM (extracellular matrix [39g]) requires footpads with different reversible binding sites analogous to ECM-binding domains of human integrins [141], nonintegrin elastin-laminin ECM receptors [142], the collagen-binding domains [143, 144] in human matrix metalloproteinase-1 (MMP-1 or collagenase 1), or other variants such as the extended poly(hydroxy)proline protein/ECM anchoring domains found in Volvox [145]. The footpad tool is rotated into, or out of, an exposed position from behind a protective cowling, using countercoiled internal pull cables. A 69 micron3 nanorobot having an equivalent spherical radius of R = 2.5 microns requires a force of Ftow = 6πηRv = 518 pN and a power draw of Ptow = Ftowv = 5 pW to be towed by grapples through T = 310 K blood plasma of absolute viscosity η = 1.1 x 10-3 kg/m-sec at a peak speed of v = 1 cm/sec [39h]. Cytoplasm and the plasma membrane has a viscosity that is 4-5 orders of magnitude higher than that of blood plasma [39i], reducing cytoplasmic transit velocity to a still-acceptable 0.1-1 micron/sec for the same nanorobot and power draw. Even using a 10-fold lower grapple number density on the chromallocyte surface (dgrapple ~ 1 grapple/micron2) than for the microbivore (dgrapple ~ 10 grapples/micron2; [42a]), a set of NgrapGC ~ 2πRdgrapple1/2 = 15 grapples positioned at equal intervals along a great circle of radius R (representing spherical nanorobot passage through a membrane surface) with each grapple able to deliver up to Fgrapple ~ 1000 pN could give an aggregate towing force of FgrappleNgrapGC ~ 15,000 pN ~ 30Ftow, or 30-fold more than the minimum requirement, greatly exceeding the desired customary tenfold redundancy design objective. Operating 15 grapples simultaneously requires only 9 pW of power. However, to ensure a wide selection and ready availability of handholds during ECM brachiation and elsewhere during the CRT mission, the chromallocyte hull incorporates grapples at about the same number density as the microbivore (~10 grapples/micron2), giving a total of 1027 grapples per chromallocyte, and includes a variety of grapple end-effectors. Although the grapples are relatively short (250 nm) when compared to nanorobot dimensions (3000-5000 nm), they should be adequate for walking on vascular and cellular surfaces whose cell plasma membranes are covered with a glycocalyx (fuzzy coat of glycoprotein strands) typically ranging from 10-100 nm thick in human cells (e.g., ~6-10 nm for red cells, 30-60 nm for bladder cells, 40-70 nm for lymphocytes, 50 nm for myocardial cells, 90 nm for cochlear hair cells) with intestinal epithelial cells having the most prominent 150 nm glycocali consisting primarily of oligosaccharide chains [39j]. The grapples should also prove adequate for vascular wall penetration (i.e., diapedesis [39k]) and the penetration of cell or nuclear membrane, since molecular handholds are plentiful. In the case of histonatation [39cc] (“tissue swimming”) and ECM brachiation [39g] through acellular tissue spaces, ECM fibrous components are typically spaced up to 10-100 microns apart [39g]. A brachiating nanorobot can pull itself along individual fibrils, changing direction at fibril junctions, indirectly working its way toward its cellular target crudely analogous to the path of a sailboat tacking into the wind. If a nanorobot happened to become entirely detached from all fibrous moorings and isolated in a liquid volume lacking handholds, two mobility alternatives are available. First, the Proboscis can be deployed to search for new handholds within a ~4 micron hemispherical work envelope. Second, the grapples may be operated as cilia, producing slow swimming motility in the fluid. Grapples can be extended or retracted in 0.25 millisec, easily allowing execution of a 2 KHz beating motion similar to that of natural cilia [39m] (e.g., paramecium cilia are ~20 times longer but also ~20 times less numerous per unit area than for chromallocyte grapples, two factors which offset because ciliary force ~L [39h]; observed paramecium propulsion velocities are 0.2-2.5 mm/sec [39n]). Unlike natural cilia, grapples may be shortened or lengthened during each stroke, and variable-area end-effectors [39xx] may also be used to enhance the propulsive effect. Note that a grapple tip speed of 1 cm/sec cycling through a 0.25 micron path length is consistent with a 20 KHz frequency of operation.
3.6 Power Supply In the microbivore design [42e], power was provided by an oxyglucose fuel cell system that required 4.453 micron3 of internal tankage, sorting rotors and machinery and 8.6 micron2 of hull surface in sorting rotors to guarantee a maximum output of 200 pW with tenfold redundancy at a power density of 23 pW/micron2 or 45 pW/micron3. In the case of the chromallocyte, minimizing nanorobot volume is a primary design criterion. Additionally, there is only limited availability of oxygen, glucose, and other energy supplies inside cells for a nanorobot having an extended-duration in cyto mission with a relatively large total energy requirement to complete the mission. For these reasons, chromallocyte power is provided non-chemically by ten acoustic power receivers spaced at equal intervals around the equatorial perimeter of the device. Each power receiver has a piston throw volume of ~0.1 micron3 and can receive up to 200 pW across short path lengths parallel to the midsagittal plane in an operating-table scenario in which the patient is well-coupled to a medically-safe 1000 W/m2 0.5 MHz ultrasound transverse-plane-wave transmitter throughout the procedure [39t]. Each 0.1 micron3 piston measures 464 nm x 464 nm or 0.215 micron2 in area, hence the entire tenfold-redundant receiver subsystem requires a total of 1.1 micron3 of onboard volume and 2.37 micron2 of hull surface (allowing 10% extra for support structure), yielding 84 pW/micron2 and 182 pW/micron3 which is four times more compact than the microbivore fuel cell system. Somewhat higher incident power levels may be required in certain tissues such as bone, bowel and lung to overcome energy shadowing effects [39t]. Note that the simultaneous operation of 1012 chromallocytes at a 50-200 pW power draw dissipates ~100 watts inside the human body, below maximum conservative safe in vivo limits [39ac]. To provide a buffered power supply, the chromallocyte uses 0.2 micron3 diamondoid flywheels [39u] with energy storage density ~5000 pJ/micron3 that can store 5 seconds of maximum normal power draw at 200 pW. The buffering system includes 10 identical flywheels to provide the customary tenfold redundancy. A total length of ~1000 micron (~200 Zint nanorobot lengths) of diamondoid power distribution cables of radius 5 nm, each capable of carrying 1000 pW of AC power at up to 60 KHz [39v], has a total volume of ~0.1 micron3.
3.7 Onboard Computers Onboard computation and control is provided by a computer and data storage system similar to that employed in the microbivore [42b]. This includes a tenfold redundant 0.01 micron3 CPU throttled back to a ~1 megaflop processing rate to conserve energy, giving a total computer volume of 0.1 micron3, and a tenfold redundant mass memory system that is ten times larger for the chromallocyte (50 megabits, 0.01 micron3) than for the microbivore (5 megabits, 0.001 micron3), giving a total data storage volume of 0.1 micron3. The increased memory allocation is justified by (1) the increased complexity of a CRT mission as compared to an antimicrobial mission, and (2) the need for greater reliability, safety, and certainty of result in the case of CRT, where a mission failure could have more serious medical consequences. Chromallocytes can receive from the physician via acoustic signaling various parameter changes and high-level instructions while in vivo, but the nanorobots operate semi-autonomously during most of the mission.
3.8 Summary of Primary and Support Subsystem Scaling Good navigational facilities are essential for guiding each chromallocyte to its intended target cell. To this end, each nanorobot possesses ten 0.5-micron3 acoustic receivers operating in the 10-100 MHz frequency range that can intercept low-power acoustic navigational information provided by an internal navigational network [39w] or by other means [39x]. These receivers may also be operated as active transmitters for outmessaging (communication from an in vivo nanorobot to the physician), though only in buffer-powered <1 second ~1000-pW bursts due to the large power requirement [39y]. Chromallocytes can issue brief progress reports to the attending physician through the network in this manner. Each receiver is allocated an additional 0.1 micron3 of support structure. Duty cycles of both receivers and transmitters for both power and information signals are less than 100% and each function may be allocated fixed or optional temporal windows in each clock cycle to avoid overlap. The chromallocyte is allotted a quantity of internal and external chemical, pressure, and temperature sensors equivalent to twice the numbers used in the microbivore [42c], since the chromallocyte is larger and also needs additional external sensors to allow (1) identification of the many different local environments through which it must pass (e.g., bloodstream, vascular wall, ECM, cell plasma membrane, cytoplasm, nucleoplasm), (2) quick and convenient acquisition of handholds for the mobility system, and (3) validation of the identity of encountered tissue types. This will include ~2000 external chemical sensors (sensor dimensions 10 x 45 x 45 nm with 450 nm2 face area) and perhaps several hundred additional external sensors of similar size to measure temperature, perpendicular and shear contact pressures, and local tangential fluid velocities. Onboard tankage, surface sorting rotor pumps, external ports, internal pumps and piping for chemical consumables is also allocated 1.2 micron3 and 0.5 micron2 of hull area in the baseline design, which includes 1 micron3 of reagent storage. Many of these support systems are located in the equatorial “utility shelf” volume indicated in Figure 2. The chromallocyte must also discharge and reacquire many chemical substances such as inhibitors and enzymes during the mission. The design includes 1000 sorting rotors (e.g., Section 5.2, Step 20) for each of 20 different chemicals; applying the usual tenfold redundancy gives a total of 200,000 sorting rotor pumps occupying 2.8 micron2 of hull space and <0.2 micron3 of device volume. An additional 1.474 micron3 of unspecified mechanisms and support structure is included in the current baseline design, which is summarized in Table 2.
Note that if 100% physical compaction of chromatin could be achieved, or alternatively if chromatin protein content could be entirely eliminated from the removal volume and new (protein-free) DNA to be implanted was still physically compacted only to ~50% for transport inside the chromallocyte, then the total required onboard vault volume would shrink from 40 micron3 to 20 micron3, reducing optimal chromallocyte dimensions to 3.3 x 2.6 x 5.0 microns (~43 micron3) using the same design schema. In the most extreme case, one might imagine eliminating all transportable protein and all introns (traditionally assumed to be “noncoding” DNA) from the manufactured installable chromosomes if this could be done without any negative medical consequences, though intron deletion appears problematic because many introns have important transcriptional promotion, structural, and other epigenetic cellular functions [146]. Such severe genomic compaction would reduce transport requirements to just a ~0.3 micron3 exon-only genomic payload retaining just ~3% of the original genome length, which might allow the chromallocyte to be redesigned using a “digest and discharge” [39z] (and then “replace”) schema yielding a nanorobot architecture more closely resembling the microbivore [42] in size and capabilities.
4. Ex Vivo Chromosome Sequencing and Manufacturing Facility
To manufacture the replacement chromosome set that will be transported to a specific cell by a chromallocyte, the patient’s existing cellular chromosome set must be obtained and sequenced using a fast ex vivo DNA reading facility, after which the sequence data can be passed to a DNA fabrication facility that can manufacture the new chromosomes and pack them into the chromallocyte.
4.1 Genome Sampling and Modification In order to replace the patient’s existing genome in a CRT procedure, appropriate DNA samples must first be obtained. The exemplar whole-organ CRT mission will customarily involve chromosomal replacement in organs which may have as many as 5-10 different cell types present. In the future nanomedical treatment environment in which CRT would be practiced, templates for the standard human DNA sequences in each organ and cytotype (including organ-specific epigenetic information such as methylation patterns) will be readily available. The task for the CRT practitioner is to ascertain how the patient’s personal DNA – including the >1 million known single nucleotide polymorphisms (SNPs) [147] and other structural and epigenetic variations (see below) – differs from standard sequences by directly sampling the patient’s genome. Genome acquisition would begin with a microbiopsy of the organ that is intended to receive CRT. This could be done using either micron-scale painless skin-penetrating reticulating tissue probes or a small set of chromallocyte-class nanorobots designed for chromatin-nondestructive DNA extraction. In this process, all nuclear DNA would be removed intact from 100 sample tissue cells from each cell type in the target organ, collected from well-separated locations throughout the organ. If desired, the nuclei of the 100 sampled cells could simultaneously receive a temporary generic maintenance genome to absolutely ensure cell viability for some number of hours until they can receive CRT (along with all other cells in the target organ) from the chromallocytes that will be administered later in the procedure, which could be inserted using a procedure similar to the one described in Section 5.2. However, normally the destruction of ~0.000001% of randomly-placed sampling cells should not disturb overall organ function in any way. The DNA collected from each set of 100 cell nuclei is sequenced (see below) and compared. This yields a consensus sequence that should be identical to the original pristine fetal DNA such that all sequence information errors due to DNA damage accumulated during the patient’s lifetime (e.g., due to aging) have been averaged out. The statistical probability that a majority of 100 independent DNA samples of each cytotype will possess identical random single base pair errors at the exact same positions in the sequence is vanishingly small. Note that the consensus sequence for each chromosome will include one maternal- and one paternal-contributed set. Systematic base pair errors and variations are more problematic. For example, all descendents of a reproducing cell that bears a retrovirus-modified sequence will retain the same sequence modifications, but this will be a known effect of the retrovirus and hence recognizable and correctable. Additional mixing may occur with recombination or transposons (see below), but not identically in all or a majority of cells, hence is excludable. Some regions of the genome (e.g., hypervariable minisatellite DNA repeats at recombination hotspots [148, 149], hotspots at microRNA genes [150], and the hypermutating immunoglobulin variable region (Section 6.7) and proto-oncogenes [151]) may be more susceptible to recombination or mutation [152] than other regions and hence might accumulate similar types of errors preferentially in those locations. To deal with such systematic errors, the consensus sequence should be compared to earlier genome sequencing scans that may be recorded in the patient’s medical records, perhaps even from infancy, and these may be augmented by further sampling, either from the patient’s quiescent stem cells [153] (very low-activity cells likely to retain the most pristine copies of the “original” genome) or from the patient’s undifferentiated white blood cell (WBC) [154], hepatic [155], muscle [156], neural [157] or other progenitor cells which have presumably undergone relatively fewer mutations and may be more abundant than stem cells. Comparing even a few of these will allow all errors other than those in the original fertilized egg cell to be detected and eliminated. Additionally, the unequal recombination of numerous (noncoding) minisatellite DNA regions (tandemly repeated 10-100 bp units typically representing <10% of mammalian genomes [87]) can slightly adjust region lengths, making every individual genome distinct [87], but these systematic variations should be easily recognizable in the sequence data. Comparison to a few representative sample genomes from other organs of the patient might be warranted to detect any possible epigenetic organ-specific asymmetric distribution patterns of post-mitotic old/new sister chromatid copies, as has been proposed by Armakolas and Klar [158]. Another complication is the discovery [159] that in addition to single nucleotide polymorphisms, the genome of each person also has natural genetic “structural variations,” including most importantly deletions, duplications and large-scale copy-number variants – collectively termed copy-number variants or copy-number polymorphisms [160] – as well as insertions, inversions and translocations [161]. There are 1,447 copy number variable regions (CNVRs) in the human genome [162], ranging in size from 960 bp to 3.4 Mb [163], with ~12% of the genome variable in copy number [162]. These structural variants can comprise millions of nucleotides of heterogeneity within every genome, and possibly may contribute to human diversity and disease susceptibility [161]. An initial mapping [164] has located 415,436 unique human insertion and deletion (INDEL) polymorphisms, ranging from 1-9989 bp in length and split almost equally between insertions and deletions. The map identifies five major classes of INDELs: (1) insertions and deletions of single-base pairs, (2) monomeric base pair expansions, (3) multi-base pair expansions of 2-15 bp repeat units, (4) transposon insertions, and (5) INDELs containing random DNA sequences. These INDELs are distributed throughout the human genome with an average density of one INDEL per 7.2 kb of DNA [164]. Since all cells in a target organ of a given patient should share the same structural variants, the patient’s uniquely variant genomic structure should be reconstructable during sequencing based on information provided by the CRT genomic sampling procedure previously described. Additional complications involve cell differences due to epigenetic gene regulation, most notably methylation (e.g., only ~10% of genes are activated at any given time. The eukaryotic genome is normally demethylated, then re-methylated early in embryonic development via epigenetic reprogramming [165]. After birth, differences in maternal care can induce differential methylation patterns in some promoter regions, influencing gene expression [166]. Genome-wide hypomethylation [167] and hypermethylation [168] may be implicated in cancer. An important special case is X-chromosome inactivation (XCI) [169] in which the X-chromosome contributed from either maternal or paternal parent is silenced (becoming Barr bodies [170]), leaving the other one active, in female mammalian cells. XCI for each cell is chosen randomly [171] during embryogenesis and is presumed to be a permanent selection for all descendants of a cell. While X-linked transmembrane proteins such as the teneurins [172] reside on the plasma membrane and thus may be antigenically visible, even in rare cases of extremely skewed XCI (e.g., 80% of cells favoring one parent [173], rather than the usual ~50%), the immune system recognizes both isoforms so a different assignment to any particular cell during CRT should elicit no immune response. Nevertheless, a conservative protocol for CRT would ensure that any replacement genome will encode the parentally-correct XCI for each cell that is subject to treatment. The XCI information may be acquired for each cell during initial whole-organ mapping by using chemical sensors to examine appropriate plasma membrane proteins such as teneurin and then to record whether the maternal or paternal variants are expressed in that cell. The patient’s genome can be repaired or modified during the replacement process. At minimum, the replacement set of manufactured chromosomes will be defect-free copies of the originals from which a lifetime accumulation of aging- [174], free-radical- [175], and genotoxic chemical- [176], bacterial- [177], viral- [178] and other disease-related modifications to the DNA of surviving cell lines have been removed. Telomeres in non-cancerous cells can be restored to their full length, a key modification providing effective cellular immortalization [179] as part of an anti-aging therapy [180-182]. Genetic errors causing mis-methylation as in fragile X syndrome [183] can be corrected. Short genetic sequences called viral retrotransposons [87] or endogenous retroviruses (ERVs) [184] that have been semirandomly inserted into the genome by retroviruses during the patient’s lifetime can be readily recognized by comparison to identical or similar sequences recorded in the publicly available database of known retroviral sequences, and thus can be edited out of the consensus sequence if desired. Additional post-sequencing modifications to the patient’s original genome can also be done prior to fabrication of the new chromatin. For example, inherited genetic diseases may be eliminated by replacing the affected gene sequences with new sequences drawn from a databank of statistically “normal” sequences for the patient’s particular gender, race, ethnic background, and MHC-related tissue type. In the case of point mutations (e.g., sickle cell disease, caused by a single base pair substitution), such corrections are less likely to affect the activity of other genes. Tissue-resident cancerous cells that the physician would prefer for structural reasons not to remove (e.g., via microbivore-class nanorobotic digestion [42] or pharmacyte-induced apoptosis [44]) but to leave physically intact in situ would automatically be reprogrammed with the “normal” genetic complement for a cell of that type due to the generic nature of the CRT process, likely reversing the pathology. Additional genomic housecleaning may include moving nonparasitic transposons (“jumping genes”) from their current chromosomal location back to a statistically normal chromosomal location, or alternatively to a new chromosomal location that can be shown to provide improved cellular (e.g., regulatory, transcriptional, etc.) function or long-term genomic stability. Transposon-derived repeats constitute up to ~50% of the human genome [185]. Minisatellite DNA segments can be regularized in length, or eliminated (for unstable minisatellite segments such as CEB1 in humans [186]), as can the purely parasitic transposons. Common transposable genetic sequences found in primate DNA [187] are mostly of two types: LINEs (long interspersed sequences, typically ~6500 bp long, now believed to be of viral origin) and SINEs (short interspersed sequences). Primate genomes contain, for example, 1200-2000 copies of a LINE retrotransposon called L1 [188]. LINEs may not be entirely benign. For instance, L1 elements in cultured human cancer cells were found to delete DNA 10% of the time when they jumped to a new location, possibly knocking out genes or creating devastating mutations in the process [189, 190]. In the human genome, the most common SINE is the Alu family, whose members incorporate similar repetitive sequences of ~300 bp that are interspersed with nonrepetitive DNA in ~300,000 copies in the haploid genome, equivalent to one copy per every 6000 bp of code [87]. If definitively proven to be transcriptionally and epigenetically inactive, these repeat sequences might be eliminated even though their contribution to the incidence of cancer may be relatively small [191] and even though they might be essentially neutral residents of the human genome [192]. Less conservatively, they could be replaced with unique sequences that could contribute to genomic stability by minimizing defects that can develop due to improper homologous recombination repair of DNA [R. Bradbury, personal communication, August 2002]. (Bradbury notes that the homologous recombination repair pathway [193, 194] must search for a DNA strand that is “similar” to the one that is broken. If the strand break occurs in a transposon sequence, the repair system is likely to find a random similar sequence from which to do the repair that is not the precise sequence it should be using on the chromosome that is homologous to the broken one – a possible contributing factor to both cancer and aging.) However, Alu sequences may serve important functions given that: (a) the presence of Alu repeats is responsible for at least some RNA secondary structure [195, 196], (b) translated Alu sequences can assist in nuclear localization [197], and (c) truncated Alu sequences may be implicated in oncogene suppression [198] and in human T-cell silencing [199]. This argues against Alu deletion. Beyond repetitive sequences, it might also be possible to shorten or eliminate many introns and even some duplicative exons not expressed in a particular cytotype or cell lineage, effectively compressing the human genome into a more compact code that could make cell reproduction faster or more efficient. But the effects of (a) extensive genomic compression [200] (e.g., upon regulated chromatin anchoring sites involving repetitive DNA that might control gene expression [201]), possibly including intron removal [202, 203], and (b) capability augmentations (e.g., addition of endogenous whole-genome surveillance systems [204]) remain unknown and further analysis of such options (and their advisability) is beyond the scope of this paper. For instance, “junk” DNA may be needed to position exons so that splicing takes place correctly, or to serve as “sinks” or “decoys” for mutagens to reduce the likelihood that a transcriptionally important gene is corrupted (rather than the “junk” that is less important), and other functions are also likely. In all these cases, care must be taken to ensure that new protein variants are not introduced that could trigger an immune system response. For instance, 20-25% of the human population carries the apolipoprotein E4 (ApoE4) allele, a gene which is associated with an increased risk of developing Alzheimer’s disease; the other two more common ApoE isoforms, the ApoE2 and ApoE3 alleles, lack this predisposition [205-207]. Replacing the ApoE4 allele in a heterozygous patient (i.e., an ApoE2/3 allele on one dsDNA strand and an ApoE4 allele on the other homologous strand of the diploid chromosome) is safe because both proteins are normally present in the body, hence both will be properly recognized as “self”. But replacing both ApoE4 alleles in a homozygous patient (i.e., ApoE4 present on both strands) with ApoE2/3 on both strands introduces a new protein into the body, requiring the physician to investigate whether the regions of the new protein that contain different amino acids can be displayed by the patient’s MHC alleles – which might provoke an immune system foreign body response.
4.2 Chromosome Sequencing A nanorobotic DNA sequencing facility could be extremely small, fast, and cheap. For instance, J. Craig Venter has been explicitly pursuing the goal of $1000/copy human whole-genome sequencing by 2012 [208]. In 2003 the J. Craig Venter Science Foundation offered a $500,000 prize for achieving this milestone, then announced in 2005 that it was seeking to raise the prize to $10 million [209]. The NIH has also solicited proposals for research leading to the $1000 genome [210]. Notes Venter [209]: “Once this threshold has been reached, it will be feasible for the majority of individuals to have their genome sequenced and encoded as part of their medical record.” The 1G Analyzer instrument manufactured by Solexa (http://www.solexa.com, recently merged with Illumina http://www.illumina.com) is estimated to be able to produce a human genome sequence for ~$100,000/copy in ~3 weeks. Pushing whole-genome sequencing costs down to $1000 or below may require a nanotechnology based approach. As a preliminary effort along these lines, Branton’s team at Harvard University has conducted an ongoing series of experiments using an electric field to drive a variety of RNA and DNA polymers through the central nanopore of an alpha-hemolysin protein channel mounted in a lipid bilayer similar to the plasma membrane of a living cell [211, 212]. As early as 1996, these researchers had determined that the individual nucleotides comprising the polynucleotide strands must be passing single-file through the 2.6 nanometer-wide nanopore, and that changes in ionic current could be used to measure polymer length. By 1998, Branton had shown that the nanopore could be used to rapidly discriminate between pyrimidine and purine segments (the two types of nucleotide bases) along a single RNA molecule. In 2000 came demonstration of the ability to distinguish between DNA chains of similar length and composition that differ only in base pair sequence, and Branton continues to perfect this approach [213-218]. Current research is directed toward reliably fabricating pores with specific diameters and repeatable geometries at high precision [219-221], understanding the unzipping of double-stranded DNA as one strand is pulled through the pore [222] and the recognition of folded DNA molecules passing through the pore [217], identifying DNA phosphorylation patterns [218], performing experiments with new 3-10 nm silicon-nitride nanopores [217], and investigating the benefits of adding electrically conducting electrodes to pores to improve longitudinal resolution “possibly to the single-base level for DNA” [217]. It has been estimated [223] that nanopore-based DNA-sequencing devices could allow per-pore read rates potentially up to 1000 bases per second. A reader comprising 1000 nanopore DNA-reader sites each reading at 1000 bp/sec could in principle sequence the entire human genome in ~1 hour, a conclusion supported by recent computational simulations by Lagerqvist et al [224]. The typical 24-hr replicative human cell cycle time (Section 6.1) also supports the idea that reading the whole genome can be done very rapidly, even by biology. Mature nanomechanical sequencing systems should allow the speed of DNA reading to be improved by another 1-2 orders of magnitude beyond these diffusion-based methods, as illustrated by the following simple scaling analysis. We start by placing a whole-chromosome sample from a single cell in a small fluid-filled sorting chamber which is perhaps a few microns wide. Histones, other proteins, and attached mRNA strands are enzymatically removed and discarded, then the chromosomes are biochemically and mechanically separated into 46 haploid strands of dsDNA having a total duplex-DNA contour length of ~2 meters. A length measurement and a few other simple tests determine the chromosome number of every double strand. Each dsDNA duplex strand, ranging in length from 15.7-82.1 mm in contour length and suspended in an appropriate carrier fluid, is then transferred into a separate long narrow channel. The positionally-controlled end-effectors [39aa] of two nanorobotic manipulator arms [39bb] attach to either end of the dsDNA to establish orientation. The strand is cleaved in half at the center of the strand using a nuclease-like enzymatic end-effector mounted on a third positionally-controlled manipulator arm or sharp scanning probe tip [225-229], and the two cleaved ends are attached [230] to another pair of manipulator arms to maintain the two strands in a known orientation. (Cleavage of individual chromosomes via femtolaser nanosurgery was first demonstrated experimentally in 1999 [231].) Each half is transported under full positional control to a second pair of manipulator arms where the cleaving process is repeated. After 10 such cleavings, there would be a total of 1000 strand segments each of length 15.7-82.1 micron (47,000-245,000 bp) rigidly held by ~2000 manipulator arms. Each strand segment would then be mechanically scanned using an AFM-mounted tooltip that can chemotactically sense the size and orientation of the exposed nucleotide bases comprising each base pair, along with their methylation, thus fully sequencing the segment [232-234]. Alternatively, dsDNA strands can be mechanically unzipped by applying as little as ~15 pN of force [235] and it may be possible to partially infer single base-pair sequence by this means because the force is base-pair dependent [236-239]. (Contemporary scanning probe techniques are more commonly used to produce 3D ultrastructure images of chromatin strands with a minimum feature resolution of some tens of nanometers [226-229].) If: (1) initial sample preparation in the sorting chamber requires 10 seconds, (2) fluid transfer into the first channel occurs at ~10 mm/sec over a maximum 82.1 mm distance, (3) each subsequent positionally-controlled cleaving, attachment and transfer operation takes ~5 seconds, given that a manipulator end effector requires only 0.1 sec to be transported 100 microns at a conservative arm velocity of 1 mm/sec, and (4) the linear nucleotide sequencing of the final segment occurs at a conservative net scan rate of 1 micron/sec, then the total throughput time to mechanically sequence the longest chromosome would be ~150 sec. A contemporaneous scanning process could measure the methylation pattern of the dsDNA sample segments, matching these to the by-then-known human methylome [240-242] for this cytotype and organ. If: (1) the volume of each channel is ~1000 times the volume of the dsDNA segment placed within it, giving ~100,000 micron3 of total channel volume for each haploid strand sequenced, and (2) the 2000 manipulator arms including all appurtenant structures each averages ~1000 micron3 in volume, giving ~2,000,000 micron3 of total manipulator volume for each haploid strand sequenced, then the volume of channels and manipulators to process all 46 haploid chromosomes from a single cell sample, in parallel, is ~0.1 mm3. Simultaneously processing 100 different cell samples from each of 10 different cytotypes in the target organ thus requires ~100 mm3 of nanomechanical sequencing hardware with a parallel total throughput time of ~150 seconds for all 1000 cell samples. Total power requirement for simultaneously operating 2000 x 46 x 1000 ~ 108 manipulator arms assuming ~10 pW per micron-length manipulator is ~1 mW, a modest ~104 W/m2 power density overall.
4.3 Chromatin Synthesis How fast can chromatin be manufactured? Scaling analyses [51b, 243, 244] for convergent-assembly-based desktop-sized nanofactories for manufacturing molecularly precise diamondoid products using positionally-controlled mechanosynthesis predict a throughput rate of ~100 sec with productivity ratios of ~1 kg/hour of product per kg of nanofactory. Analogously in biology, ribosomes assemble proteins according to digitally encoded instructions using mechanochemically-driven positionally-controlled placement of amino acids in vivo [245]; ~1 kg of proteins can be assembled by 1 kg of bacterial ribosomes in 270-710 sec, or ~1 hour for eukaryotic ribosomes [244]. Biological mechanochemistry is employed in DNA-related systems [246] and the mechanochemistry of organic polymers using positionally-controlled scanning probe microscope tips has been demonstrated experimentally in a liquid solvent environment [247]. Alternatively, DNA could be assembled as a rigid polymer structure at low temperature (20-80 K) and in vacuum using mechanosynthetic tooltips similar to those envisioned for the fabrication of diamondoid structures under similar conditions [248-250]. We assume that DNA and protein assembly times in nanofactories specialized for this purpose may be comparable to the assembly times required to manufacture diamondoid products, given that the making and breaking of predominantly C-C, C-H, C-N and related covalent bonds are involved in both cases. Hence ~1012 copies (the maximum for the largest organ) of the entire chromatin (DNA + protein) mass of ~2.93 x 10-11 gm per cell (Section 2.2) requires the manufacture of ~29.3 gm of genetic material and should take ~100 sec, roughly matching the continuous throughput time. Note that custom DNA and peptide sequences have been available for online purchase for many years [251]. Whole gene synthesis has been actively pursued throughout the 1970s [252-254], 1980s [255-258], 1990s [259-261] and 2000s [262-267]. By early 2000, Glen Evans was the first to produce made-to-order DNA strands up to 10,000 nucleotide bases in length [268] and was striving to increase this length by at least a factor of ten. In 2003, Venter’s group at IBEA reported synthesizing the complete genome of a small (5386 base pairs) phi X bacteriophage virus in just 14 days [269], and Egea Biosciences’ proprietary GeneWriter™ technology produced the then-largest gene that had ever been chemically synthesized – over 16,000 bases with a claimed base-placement error of only ~10-4 [270]. By 2004 a University of Houston group had assembled 10,000-base-pair DNA constructs [271], a group at Children’s Research Institute reported synthesizing two 20,000 base pair products [272], and a group at Kosan Biosciences had synthesized a functional 31,656 base pair polyketide synthase gene cluster [273]. A group at Vanderbilt University is planning the synthesis of a 113,000 base pair minimal genome containing 151 genes [274], probably long enough for a very simple bacterial genome. Synthetic oligonucleotides have also been enzymatically methylated [275, 276]. With synthesizable genomes increasing in length ~100-fold per decade since the 1980s, a simple log-linear projection would indicate the probable availability of whole human genome synthesis by the 2020s. However, positionally-controlled molecular manufacturing would represent a fundamentally different and still more advanced process compared to present-day oligonucleotide hybridization and DNA assembly techniques, possibly avoiding many of the current limitations of those techniques such as high cost, low speed, and high error rate [277-279] that produce unplanned process mutagenicity. The manufactured genetic material destined for each target cell will consist first of manufactured duplex dsDNA which is assembled [280] from two complementary strands of ssDNA that have been positionally (i.e., non-diffusionally [281]) fabricated base by base, in the desktop nanofactory [52]. Using positionally controlled functionalized tooltips analogous to DNA methyltransferase enzymes [282-284], the dsDNA haploid strand is next partially methylated for the particular cell type, thus allowing the DNA, once installed in the target cell, to express only the appropriate exons (at non-methylated sites) that are active in the cell type to which the nanorobot has been targeted. (Methylation involves the addition of methyl groups to deactivate expression of a section of DNA.) The appropriate X-chromosome strand in each female genome is silenced. The partially methylated strand is then wrapped around properly modified [285] (possibly acetylated [286], methylated [287], phosphorylated [288], monoubiquitylated [289], or sumoylated [290]) histones – incorporating post-translational modifications perhaps constituting a “histone code” that may be used by the cell to encrypt various chromatin conformations and gene expression states [291] – after being joined with its homologue strand to make a diploid chromosome that is joined at the waist by a centromere. (Further research may determine if alternative processes involving partial self-assembly of histones [292] and other chromatin-associated proteins [293] onto positionally synthesized DNA can be equally reliable but more efficient.) The centromere is coded by a conserved relatively short repeating heterochromatin sequence and includes a complex of associated proteins [294]. Each completed 23-diploid-chromatid set of chromatin fiber would be fully condensed (e.g., by nondisruptive methods functionally similar to chemically-induced premature chromosome condensation [295]) into compact supercoiled form [296, 297], then be mechanically packed [298, 299] into one chromallocyte device that will be sent to a single target cell to replace the natural genetic material in that cell nucleus. Such sets of chromosomes could also be packaged into isolated nuclear membranes to form complete artificial nuclei, which may be useful in other missions or applications. Some additional hardware and time will be required to pack the chromallocytes with the DNA-related material, recharge the nanorobots with consumables, and download appropriate software and specific mission parameters into the onboard chromallocyte nanocomputers. One trillion chromallocyte preparation stations, each allotted 10 times the nanomachinery volume of a single nanorobot and each able to prepare chromallocytes at 0.0005 Hz (consistent with the 1950 sec single-vault pumping rate estimated in Section 3.4), would occupy a total volume of ~700 cm3 for all 1012 stations and could collectively prepare 1012 nanorobots for a single-organ CRT procedure in ~2000 seconds with a continuous power draw of at least ~26 watts for fluid pumping. (More efficient nanorobot loading procedures can be envisioned that could significantly reduce these time and energy estimates, e.g., a removable rear end plate providing direct vault access, but either parameter could be several orders of magnitude larger without jeopardizing CRT feasibility.) The resulting exemplar combined chromosome sequencing and manufacturing facility is envisioned as a desktop appliance which accepts, as inputs, patient cell samples for rapid genomic sequencing, empty premanufactured chromallocytes, DNA precursor chemicals and other nanorobot consumables, along with the physician’s directives and mission design, and then produces, as its output, 10-100 cm3 batches of fully-loaded programmed chromallocytes ready for injection into the patient, in total processing times on the order of ~1 hour. Genetic material mechanically loaded into a chromallocyte at viral packing densities and pressures is likely to have a long undamaged shelf life [300], given that freeze-dried samples of variola (smallpox) virus are still infectious after storage for 20 years [301] and samples have been shown to retain their virulence after storage in ambient conditions at room temperature for at least 18 months [302] and in one case up to 13 years [303].
5. Mission Description This Section describes a sequence of events likely to occur during a typical chromallocyte mission in which all cells in a specific organ inside the human body have their chromosomes replaced with new genetic material. In this procedure, the patient is scanned and prepped while a dose of personalized therapeutic chromallocytes is manufactured. After infusion into the patient, these mobile cell-repair nanorobots perform limited vascular surface travel into the capillary bed of the targeted tissue or organ. This is followed by extravasation, histonatation, cytopenetration, and complete chromatin replacement in the nucleus of the target cell, ending with a return to the bloodstream via the same route and subsequent extraction of the devices from the body at the original infusion site.
5.1 Mission Summary The entire single-organ CRT procedure should be completed in an estimated ~7 hours, including patient preparation and post-operative activities but excluding rescheduled uncompleted activities. CRT could be performed in five phases as follows: (I) Organ Survey. Install an in vivo nanorobotic mobile navigation grid [39w] in and around the patient’s target organ if one is not already present, then use similar navicyte-class nanorobots [39w] to map the organ and its adjacent tissues and vasculature to micron-scale resolution. Compile comprehensive list of cytotypes, cell cycle status (Section 6.1), epigenetic status (e.g., X-inactivation; Section 4.1), and cellular addresses for cells intended to receive CRT in the target organ. Required time ~1 hour. (II) Chromallocyte Preparation. Collect samples of patient’s DNA from target organ for all cytotypes (Section 4.1). Analyze DNA sequence and make necessary adjustments, repairs or modifications to the genetic sequence (Section 4.2). Manufacture new DNA, load DNA into ~1012 chromallocytes and program them, each targeted to a specific individual cell (Section 4.3). Required time ~1 hour. (III) Patient Preparation. Place patient on ultrasonic vibrating table with comfortable encapsulated gel interface to maximize acoustic power transmission into the body, providing energy for in vivo chromallocyte activities. Sedate the patient while simultaneously injecting respirocytes (nanorobotic oxygen carriers [41]) or employing hypothermia to allow much reduced pulse rate and greatly slowed blood velocity. Install self-guiding flexible nanocannula directly into blood vessel nearest to the target organ through which chromallocytes can be conducted into the patient’s body, and through which excess fluid can be removed as required. Required time ~0.5 hour. (IV) Chromosome Replacement. Introduce through nanocannula up to ~1012 chromallocytes (one per target nucleus) having a maximum nanorobot displacement volume of 69.250 cm3 (Section 3.1), a 1 terabot (trillion device) dosage comprising a modest 7% volume concentration of nanorobots when suspended in 1 liter of saline carrier fluid. This fluid also may contain recharged respirocytes [41] to maintain proper tissue oxygenation, microbivore [42]-class devices to help maintain an aseptic vascular environment, and limited volumes of other auxiliary nanorobots for specialized purposes. Each chromallocyte requires a total of ~3 hours to travel to the target organ, complete its tasks in its assigned target cell in that organ, and then return to the entry point, as detailed in Section 5.2. All chromallocytes are infused into the patient in ~1 hour; after 3 hours, the first-infused nanorobots have completed their CRT activities and begin to exit through the nanocannula in the opposite direction, an egress that occurs over the next ~1 hour until the last-infused nanorobot has departed. Required time ~4 hours. (V) Patient Post-Operative. Restore patient’s normal blood flow and pulse rates, extract respirocytes and other auxiliary nanorobots, then uninstall the in vivo nanorobotic navigation grid if desired. Required time ~0.5 hour.
5.2 Detailed Sequence of Chromallocyte Activities The ~3 hour chromosome replacement process to be performed by each chromallocyte during Phase IV includes a 26-step sequence of distinct semi-autonomous sensor-driven activities, described below. This sequence must be regarded as illustrative only, given that, e.g., cell signaling pathways are as yet incompletely understood. (1) Injection. The nanorobots are introduced through a flexible telescoping nanocannula (similar to transdermal [304] microcannula [305, 306] but including biochemical and chemotactic nanosensors [39pp] and nanomotorized guidance) into a small blood vessel located near the target organ. The injected nanorobots ambulate along the blood vessel surface [39ee] until they reach the designated target organ. Using information obtained from the navigational grid and the previously prepared map, the nanorobots negotiate the continually bifurcating vasculature until they reach the capillary bed, and then the specific capillary vessel, that is located closest to their target cell. Each nanorobot is assigned the address of a specific cell whose relative 3D spatial location is known to within ~3 microns of accuracy [39w] on the map. For additional safety, the nanorobot uses contact chemical sensors to detect the presence of organ-specific vascular ligands [307] to verify its arrival in the correct organ. Nanorobots that cannot unambiguously and definitively locate their target cell should exit the body and report their failure to the physician who may then adjust the mission accordingly. Note that it might be possible to eliminate the in vivo navigation system and rely instead on less sophisticated means to find cells appropriate to receive payloads, or even to eliminate the precise one-to-one match between prepared chromallocytes and the cells to which they are targeted, but exploration of these and related simplifications are beyond the scope of this paper. (2) Extravasation. The nanorobots employ controlled diapedesis [39k] to penetrate the local endothelium of the capillary bed nearest to or within the target organ to gain entry into the tissues. Diapedesis by natural leukocytes [308] typically requires 3-10 minutes to complete [39k], a slow pace that may help ensure vessel wall transit without lasting damage. However, once a passage is opened, safe transit times on the order of 3-10 sec per device for a continuous convoy of mechanically powered nanorobots appear feasible [39k]. If the total vascular wall area to be breached throughout the capillary bed [39nn] of a 1 kg organ is ~4 m2 and the nanorobots pass single-file through ~14 micron2 holes taking ~10 sec per transit (~300/hr), then a 1-hr infusion of 1012 nanorobots traveling in convoy formation [40d] requires holing a modest ~1% of the vascular surface. Fluid leakage through temporary nanorobot transit gaps should not exceed, even in the most severe cases, the normal extravascular water exchange rate [40e] and all breaches are resealed at the conclusion of convoy transit [39k]. Electron microscopic studies show that an efficient protein-tight seal is maintained between endothelial cells and a migrating leukocyte during all stages of its escape [309]. (3) ECM immigration. The nanorobots proceed through the extracellular matrix (ECM), if transit through acellular tissue is required, moving toward the target cell using their mechanical mobility system (Section 3.5) for histonatation [39cc] and ECM brachiation [39g]. While leukocyte speeds through ECM are typically 0.01-1 micron/sec [39g], depending on tissue type, nanorobot brachiation through ECM up to 100 micron/sec appears to be both feasible [39g] and safe [40f], implying a vasculature-to-target-cell transit time of ~10 sec. Nanorobot passage at these speeds should not induce a biological mechanical stress response [40g]. In some cases, upon reaching densely packed cellular tissues the chromallocyte may need to transit around or through at most 1-2 intervening cells before reaching its designated target cell [39hh], a process already demonstrated in biology at smaller sizes and speeds. For instance, actin-based propulsion through the cytoplasm of one cell with entry into a neighboring cell is practiced by the spotted fever group rickettsiae [310] at speeds of ~0.07 micron/sec [311], allowing whole-cell transit in a few minutes. Nanorobots should safely transit cytosol up to ~10-100 times faster [39qq]. If mechanical disruption of intercellular adhesion contacts by passing nanorobots elicits unwanted cell responses, mechanotransduction can be temporarily inhibited as described in Step 5. (4) Cytopenetration. Upon reaching its target cell, the chromallocyte fully enters the cell by cytopenetrating through the plasma membrane [39ii] with breach-sealing behind it [39jj, 40i] and minimal leakage [39oo]. Analogously, exocytosis-based resealing of a microneedle puncture through a fibroblast plasma membrane occurs in 5-10 sec [312, 313] and a second puncture at the same site heals even faster [314]. Several mechanical cytopenetration techniques for nanorobots have been described elsewhere [39ii], but the chromallocyte might best employ a solvation wave drive [39kk] employing movable reversible semaphore binding sites [39f] located on the device surface which are assisted by the motile grapple array (Section 3.5). The slightly convex prow of the nanorobot may facilitate membrane contact and penetration. Note that a ~5 micron long device performing CRT on a 20 micron cell may reside entirely in the cytoplasm prior to nucleopenetration. However, the height of a renal tubular cell dependent on a peritubular basement membrane on the basal side and having a relatively short stretch of microvilli on the apical side may be only ~4 microns, in which case the chromallocyte may reside partly in cytoplasmic and extracellular spaces during CRT. (5) Block mechanotransduction. Mechanical deformation of the plasma membrane or nuclear envelope can transmit signals either into the nucleus, altering gene expression, or into the cytoskeleton, eliciting reaction from cell signaling pathways in the cytosolic compartment [315, 316]. For example, vascular smooth muscle cells use multiple sensing mechanisms to detect the mechanical stimulus resulting from pulse-related stretch and transduce it into intracellular signals that lead to modulations of gene expression and cellular functions including proliferation, apoptosis, migration, and remodeling [317]. Although nanorobot-applied forces are small in amplitude, brief in duration, very high-frequency and generally noncyclical, and even though somatic cell nuclear transfer (SCNT) succeeds despite much larger distortions of the cell and nuclear membranes during the nuclear transfer process than are anticipated during CRT, it is nevertheless possible that chromallocyte passage through cytoplasm could elicit a mechanosensory response which therefore should be inhibited during CRT in a conservative protocol. To accomplish blockage, the nanorobot may release into the cytosol engineered synthetic mechanotransduction inhibitors appropriate for the cytotype of the target cell. (Many examples of designed synthetic inhibitors [318-320] as well as de novo rationally engineered binding sites [321], enzymes [322], and proteins [323] have been reported in the literature.) These artificial inhibitors may be analogous in activity to blocking antibodies [324], K201 (JTV-519) [324], or methotrexate [325] in bone cells, TMB-8 in muscle cells [326], AIIB2 blocking-type monoclonal antibody in vascular endothelial cells [327], mepacrine and methyl arachidonyl fluorophosphonate ketone (AACOCF3) in renal cells [328], inhibitors of Rho-dependent kinase in fibroblasts [329], and so forth. Plasma membrane disruption is also quantitatively correlated with Fos protein levels [330]; releasing an engineered synthetic Fos expression inhibitor analogous to tetrandrine [331] or fangchinoline [332] would reduce or eliminate this response, and RNA interference (RNAi) might be used to selectively silence translation of existing cytosolic mRNA [333]. Distribution of a 10 kD engineered inhibitor molecule to 1 nanomolar concentration in the cytosol of a 20 micron cell would require ~2000 molecules occupying a storage volume of <0.0001 micron3 within the nanorobot; or ~100 molecules (<0.000002 micron3) for a similar concentration in nucleoplasm. Such molecules should also be engineered for maximum diffusional mobility [334] to facilitate their subsequent retrieval. (6) Nuclear localization. Once inside the cell, the chromallocyte exposes, at the anterior end of the device, a set of semaphores [39f, 42d] which will bind to outer nuclear membrane (ONM) surface but to no other surface inside the cell. Such semaphores may include contact-sensitive sensors comprised of receptors for the nuclear envelope localization domains of proteins analogous to transmembrane proteins found only in the ONM such as UNC-83 [335] or the nesprins [336]. Detection of these specific examples is complicated by possible cytoskeletal tethering [337], though antibody-like molecules targeting the cytoskeletal-nesprin or cytoskeletal-UNC-83 junctions may provide unique epitopes for recognition. The chromallocyte locomotes intracellularly, perhaps using microtubules as do intracellular bacteria [337a], until anterior contact with ONM is detected. Receptor binding to ONM proteins then provides temporary anchorage. One complication is that the nucleus is normally almost entirely surrounded by endoplasmic reticulum (ER), a dynamic network of interconnected membrane tubules that pervades almost the entire cytoplasm [338] and which the nanorobot will encounter before reaching the ONM. While there is slight evidence that the ER may be sensitive to mechanical membrane damage [339] or to the physical disruption of the supporting microtubule lattice [340], the integrity of the ER appears to be maintained during mitosis with little or no fragmentation and vesiculation [341]. It is also possible that mechanosensitive stretch-activated channels may indirectly induce Ca2+ release by the ER [342, 343], in which case the endoplasmic reticulum ATP Ca2+ pump could be reversibly inhibited using an engineered analog to BHQ (2,5-di-(t-butyl)-1,4-hydroquinone), CPA (cyclopiazonic acid) or thapsigargin [342] that could be released by the chromallocyte during nuclear transit, then reacquired by the nanorobot during cellular exit in Step 23. Passage through ER membranes will be mechanically similar to passage through the plasma membrane. Progress can be monitored by chemical detection of ER-specific luminal proteins called reticuloplasmins [344] that are found only in the reticuloplasm, the fluid occupying the cisternal spaces of the ER. Reversibly inducing localized disassembly and subsequent spontaneous reassembly of both ER and Golgi [345] may avoid transmembrane tunneling but is a more aggressive protocol. (7) Nucleopenetration. The inner nuclear membrane (INM) is lined and stabilized by the nuclear lamina layer, which constitutes a filamentous protein meshwork 20-80 nm deep. The chromallocyte extends grapple arms possessing tethered enzymatic end-effectors analogous to cellular protein kinase C that can phosphorylate and locally dissolve the nuclear lamina (as demonstrated by cytomegalovirus [346]), cutting a narrow slit or flap in the INM that can temporarily permit snug-fitting chromallocyte intrusion. Alternately, Vpr (an HIV viral protein) blebs nuclear membrane, eliciting transient extrusions resembling solar flares that subsequently rupture, allowing interchange of soluble components between cytoplasm and nucleoplasm [347]. Such localized nuclear envelope disturbances should not result in microtubule tearing of nuclear lamina as is observed during the ordered stepwise disassembly of nuclear envelope in a mitotic cell. The nanorobot should try to avoid applying physical forces to cytoskeletal elements in the immediate vicinity of the nucleus of a magnitude and frequency sufficient to trigger unwanted transcriptional, structural, or metabolic responses from within the nucleus [348-351], although general mechanotransduction will have already been blocked in Step 5 in a conservative mission design. Such non-mechanosensitive nuclear envelope penetration is exhibited by several microbial parasites including the tachyzoites of Toxoplasma gondii in mouse (possibly using its apical secretory organelle called the rhoptry) [352], Nucleospora salmonis, an intranuclear microsporidian parasite of marine and freshwater fish [353], and the merogonic and gamogonic stages of Eimeria parasites in the goose [354], and also by the MVM parvovirus [355] and by apoptogenically stimulated nuclear-immigrating mitochondria which may be serving as “cargo boats” transporting organelles or apoptotic proteins to their nuclear targets [356]. Contact with INM can be detected using sensors comprised of receptors for transmembrane protein domains analogous to UNC-84 [335] that are found only in the INM. Once sufficient penetration of the nuclear envelope is achieved, secure mechanical moorings between nanorobot and INM lamins, envelope matrix, and even external endoplasmic reticulum may be established. The nuclear envelope more stiffly resists distortion [357] under loads applied at the higher frequencies that nanorobots are likely to employ [40j], and is stiffer and more resilient than ER or plasma membrane [358]. (8) Block apoptosis. Chromallocyte activities that surgically remove DNA from cells resemble mechanical [359, 360] or chemical [361] injuries to chromosomes and appurtenant protein structures that can trigger cell apoptosis. The nanorobot must comprehensively block this “cell suicide” response cascade during CRT. Members of the caspase family of cysteine proteases that play key roles in the apoptotic disintegration of cellular architecture have been grouped by function into upstream apoptotic initiators (caspases 2, 8, 9, and 10), cytokine processors (caspases 1, 4, 5, 11, 12, 13, and 14), and apoptotic executioners (caspases 3, 6, and 7) that are the downstream effector proteases responsible for the proteolysis of key substrates. Caspases are present in the cell as zymogens that are activated by various extracellular stimuli, leading to apoptosis. The chromallocyte can disable the natural apoptotic cascade by releasing artificial caspase inhibitors into the nucleosol (and cytosol if needed). These inhibitory molecules would be synthetic analogs of natural inhibitors that are engineered for reversible action (e.g., via weakened affinity); post-CRT, remaining disabled caspases are naturally proteolytically recycled. Porous chromosome territories allow inhibitor proteins or protein complexes up to ~500 kD to quickly diffuse throughout the entire nucleus in <100 sec [362]. Caspase-specific inhibitors are known for the apoptotic initiators caspase-2 [363-365], caspase-8 [364-370], caspase-9 [363-365, 371-373], and caspase-10 [374, 375], for cytokine processors such as caspase-1 [363, 365] and caspase-12 [376-378], and for the apoptotic executioners caspase-3 [363-365, 379-381], caspase-6 [364, 365, 382], and caspase-7 [363, 365, 383-385]. (General caspase inhibitors also exist, such as the pancaspase inhibitor z-VAD-fmk [386].) It is prudent to simultaneously lock down multiple components of the apoptotic cascade because of the complex interactions among caspases – in some circumstances, caspase-6 can activate both caspase-8 [387] and caspase-3 [388, 389], caspase-3 can activate caspase-6 [389], and so forth. A death effector domain (DED) in the signaling molecule DEDD also localizes to the nucleus and can activate apoptosis from within the nucleus [390], but anti-DEDD antibodies have been raised [390] so inhibition should be feasible here as well. (9) Block DNA repair. Chromallocyte activities in the nucleus could be misinterpreted by natural biological systems as causing “damage”. In particular, DNA damage is sensed by a highly conserved mechanism involving the ATM/ATR protein sensor kinases in humans. These molecules aggregate at DNA lesions only seconds after a double-strand break occurs and activate signaling cascades that include the Chk protein kinases [391]. These, in turn, trigger both transcriptional and transcription-independent responses, including activation of DNA repair machinery via the p53 tumor suppressor pathway and cell-cycle arrest [391]. (If the DNA can’t be repaired, then p53 attempts to trigger apoptosis, an action which can be specifically inhibited by pifithrin-alpha [392], Z-1-117 [393], or imino-tetrahydrobenzothiazole-based analogs [394] if necessary.) The DNA damage response engages multiple levels of regulation involving at least 30 transcriptional factors, affecting not only DNA repair genes but also genes that influence protein and lipid turnover, cytoskeleton remodeling, and general stress pathways [391]. The nanorobot need not inhibit all transcriptional factors, but merely enough of them to achieve practical temporary blockage of the DNA repair response. The chromallocyte should initiate the blockade as far upstream as possible by releasing an engineered synthetic ATM inhibitor analogous in function to TRF2 [395], KU-55933 [396], or caffeine [397] in humans, or to similar sensor kinases found in plants [398] and bacteria [399], or alternatively by temporary direct sequestration of ATM onboard the nanorobot. The nanorobot can also intervene farther downstream in the cascade by releasing an engineered synthetic p53 inhibitor analogous in action to sodium orthovanadate [400] or pifithrin-alpha [401] which inhibit the DNA-binding activity of p53. (10) Block inflammation signals. Necrotic or damaged cells release signal molecules such as the chromatin-associated high mobility group box 1 (HMGB1) protein that binds with high affinity to RAGE (the receptor for advanced glycation end products) which is present on the extracellular surface of endothelial cells, smooth muscle cells, mesangial cells, mononuclear phagocytes and certain neurons and thus is a potent mediator of inflammation outside the cell [402], that must be blocked. Transport of free HMGB1 from nucleosol to cytosol is enabled by phosphorylation, and phosphorylated HMGB1 present in the cytoplasm will not re-enter the nucleus [403]. The chromallocyte should block the inflammatory action of HMGB1, either by releasing an engineered phosphatase inhibitor analogous in action to okadaic acid [403] or by releasing an engineered enzyme having specific recognition of HMGB1 and which competitively functionalizes the protein with non-phosphoryl groups at all phosphorylation sites such that nucleocytoplasmic shuttling of HMGB1 is not enabled. Competitive functionalization could also be performed by transporting the protein inside the nanorobot using HMGB1-specific sorting rotors (perhaps using a RAGE binding motif) and mechanochemically processing the molecules on a catch-and-release basis. The inhibitor is reacquired by the nanorobot at the end of CRT. (11) Deactivate transcription. DNA transcription activities in the nucleus (producing mRNA for export to cytoplasm) must be halted, although ribosomal translation of extant mRNA into protein will continue in the extranuclear compartment. Chromatin organization does not require ongoing transcription [404]. Nucleolar dissolution normally occurs during mitosis primarily as a result of: (a) the phosphorylation of RNA polymerase I transcription factors and processing components [405], (b) mitotic repression of RNA pol I synthesis that inactivates rDNA transcription occurs under the control of the cdc2-cyclin B kinase [406, 407], (c) mitotic disassembly of intranuclear splicing factor compartments (SFCs) (aka. nuclear speckles [408]) mediated by SR protein kinase-1 [409], and so forth. To compel dissolution of the nucleolus along with delocalization of other nuclear compartments and deactivation of transcription, the chromallocyte could release into the nucleoplasm appropriate engineered molecules analogous in action to reversible transcription inhibitors such as the adenosine analog 5,6-dichloro-beta-D-ribofuranosylbenzimidazole (DRB), alpha-amanitin, or actinomycin D (AMD) [410]. Cajal bodies (CBs) [411] are reservoirs of transcription factors that preferentially localize to the nucleolar periphery during interphase. They are released during nucleolar dissolution and will likely aggregate in free-floating intranuclear structures similar to mitotic CB remnants [412], since Cajal body and speckle components are observed to concentrate into large, distinct spots in transcriptionally inactive nuclei [413]. There are no reports of caspase-mediated SFC or Cajal body cleavage in the literature. (12) Detach chromatin. With apoptosis and other cell damage response pathways temporarily disabled, a condition which might be termed “cellular anesthesia,” the chromallocyte can now detach chromatin from its lamin B moorings in the nuclear lamina network on the nucleoplasmic surface of the INM. Lamin B is normally cleaved by the lamin protease enzyme caspase-6 that is part of the apoptotic cascade. This cleavage allows chromatin to detach from the nuclear envelope [414]. The nanorobot can use molecular sorting rotors to assimilate all free molecules of the synthetic caspase-6-specific inhibitor released earlier in Step 7, then release a small amount of a similarly engineered synthetic protease based on natural caspase-6 [415], causing chromatin detachment from the INM. The synthetic protease may then be reacquired and the inhibitor re-introduced into the nucleoplasm, thus re-establishing full apoptotic blockade. The synthetic protease should possess bulky laminophobic or lipophobic distal end groups to prevent easy passage through the nuclear envelope, since natural caspase-6 in the cytosol can attack elements of the cytoskeleton [416]. Care must be taken not to damage the lamins as a number of serious cellular diseases called laminopathies are associated with disturbances in the lamin protein structure [417]. Caspase-6 also cleaves chromatin-associated proteins such as SATB1 at their DNA-binding sites, detaching the protein from the bases of chromatin loop domains, mechanically untethering the domains [418]. Since lamina-associated polypeptide LAP2 is a DNA binding protein too, LAP2 proteolysis might be required for the complete detachment of chromatin from the nuclear envelope [414]. The LAP2 nucleoplasmic domain includes a cleavage site [414] for caspase-3 so the chromallocyte may need to repeat, using an engineered synthetic protease based on natural caspase-3 [419], the synthetic inhibitor/protease cycle previously described for caspase-6, or, alternatively, employ an analog to the phosphorylation/dephosphorylation cycle described in Step 19. Caspase-3 cleaves human scaffold attachment factor A [420], one of the major scaffold attachment region DNA-binding proteins of human cells thought to be involved in nuclear architecture by fastening chromatin loops to a proteinaceous nuclear skeleton, the putative nuclear matrix or scaffold. Caspase-3 cleaves other elements of the nuclear matrix such as nuclear mitotic apparatus (NuMA) protein [386], though there is also evidence for a possible noncaspase cleavage protease for NuMA [421] that might require retardation by an engineered analog to the serine protease inhibitor TPCK [422]. Caspases do not attack the support scaffold for the nuclear pore complexes which should remain almost intact [423]. However, caspase-3 does cleave DFF45/ICAD which releases active DFF40/CAD (DNA fragmentation factor, 40-kd subunit, aka. caspase-activated deoxyribonuclease (CAD) in mouse), a major apoptotic nuclease, causing DNA fragmentation [424, 425]. Simultaneous release of an engineered nuclease inhibitor analogous to aurintricarboxylic acid [426], a downstream inhibitor of DFF40, but lacking that agent’s undesired side effects [427] should prevent unwanted DNA fragmentation. Chromosome margination normally associated with karyorrhexis during apoptosis [428] or other cell damage response [429] thus should not be observed during nanorobot operations. Synthetic caspase-3 and nuclease inhibitor can later be reacquired via molecular sorting rotors and the synthetic caspase-3 inhibitor re-introduced into the nucleoplasm, re-establishing full apoptotic blockade. (13) Extend Proboscis. The Proboscis is extended out into the nuclear interior, with its tines retracted and its surfaces in nonadhesive mode. Numerous telescoping tines are extended, projecting perpendicular to the Proboscis trunk. Presentation semaphores on the external surface of the extended tines are rotated to their chromophilic position, immediately providing a large adhesive surface to which chromatin will strongly adhere (Section 3.2). (14) Rotate Proboscis. The Proboscis begins two slow co-rotations, the first a coaxial rotation that can wind the detached chromatin fibers into a bolus around the shaft of the manipulator, and the second a lateral gyration movement that forces the manipulator to sweep out a series of conical surfaces of progressively larger, then smaller, radii, methodically sweeping most of the accessible nuclear interior. In case a twist of strands extends from the tip of the Proboscis (rather than being wrapped around it), occasionally bending the manipulator into a U-shape while rotating the base axially should wind the twist around the shaft. As the outermost portion of the chromatin bolus adheres, and is drawn inward and coiled, the telescoping tines are gradually retracted, compacting the bolus and bringing chromatin into contact with the shank of the Proboscis, whose surface is then switched to chromophilic mode which further compacts the bolus. Maniotis et al [32] experimentally demonstrated in 1997 that chromatin can be mechanically withdrawn from a nucleus in approximately this manner by using a microsurgical technique to physically extract chromosomes from living cells under isotonic conditions, observing that “pulling a single nucleolus or chromosome out from interphase or mitotic cells resulted in sequential removal of the remaining nucleoli and chromosomes, interconnected by a continuous elastic thread.” Chromatin is much more resistant to stretching than to bending [430], hence the spooled chromatin is expected to wrap into a tight packed structure perhaps similar to the “spool models” investigated for DNA packaging in phage capsids [431, 432]. The Proboscis is rotated relatively slowly to minimize the torques transmitted back through the rotating arm to the nanorobot which is anchored on the nuclear wall, thus limiting shear forces applied to the nuclear membrane that might otherwise tear the nuclear wall. Spool time is ~1000 sec (Section 3.2). (15) Deploy funnel. After the Proboscis has spooled up the nuclear chromatin, the manipulator is moved to the nanorobot-centered coaxial position and all rotation stops as the funnel assembly is extended out into the nucleoplasm, surrounding and fully enclosing the ball of material (Section 3.3). Low gearing of the motors driving the irising aperture composed of annuli #A and #B coupled with a slow rotation of the Proboscis stalk (the tip of which remains outside the funnel enclosure) will enable the application of sufficient cutting force to cleave any stray chromatin strands that are attached to the bolus but remain outside of the interior void volume after the funnel has irised closed. These stray strands are subsequently digested in situ by nucleases (Step 16). After sealing the funnel, all tines on the Proboscis are retracted and its surface chromophilicity is switched off. The funnel is retracted slightly to maintain maximum compaction of the old chromatin while maintaining a watertight interface to the Proboscis surface, exposing slightly more of the manipulator tip. (16) Digest stray chromatin. To digest any stray chromatin that remains outside the funnel enclosure after spooling is complete, along with viral or other foreign DNA that may be present, the chromallocyte releases an engineered DNA nuclease that is faster but otherwise analogous in action to known cytosolic nucleases [433, 434] that can digest, for example, free single- or double-stranded circular plasmid cDNA in mammalian cytosol with a 50-90 minute half-life [433]. This activity provides conservative assurance: (a) that none of the old DNA [435] or even intact genes [436] can find their way into neighboring cells (normally prevented by p53 [437]), (b) that no duplicate DNA will remain in the target cell undergoing CRT (which could possibly produce excess gene copy or polyploidy pathologies; Section 1), and (c) that no nucleic acid-mediated inhibition of nuclear enzyme activity can occur [438]. If necessary, nucleotide digesta can be absorbed through the nucleoplasm-contacting surfaces of the nanorobot using molecular sorting rotors, transferred through internal channels to the cytoplasm-contacting surfaces, then discharged into the cytosol, thus preventing any possibility of nucleotide poisoning of the nucleus. The amount of stray chromatin to be digested should be relatively small in quiescent nonproliferating cells, hence is unlikely to elicit clinical pathologies such as Von Gierke’s disease (caused by excess purine), orotic aciduria (excess pyrimidine precursor), or cytolic acidification (since nucleotides such as dAMP are polyfunctional acids). Upon completion, the engineered DNA nuclease is reacquired by the nanorobot using molecular sorting rotors. (17) Dispense new chromatin. The new condensed chromatin (with associated proteins) is discharged into the interior of the cell nucleus through the tip of the Proboscis which lies outside of the sealed funnel assembly. Chromatin passage through a micropipette does not appear to damage it [33] and core histones do not separate from DNA even when chromatin is subjected to extreme stretching [439]. Slow funnel compaction aided by water pumping (Section 3.4) forces old waste chromatin into the vault spaces vacated by dispensed new chromatin and exposes more of the Proboscis exterior, increasing its available work envelope as discharge proceeds. The moving Proboscis places each strand near what should become its future chromosome territory (CT). Chromosomes occupy preferred nuclear locations but there is no evidence of a rigid suprachromosomal order [440]. No “north” or “south” pole of the short nuclear axis, “west” or “east” pole of the long axis, or other compass polarizations can be distinguished in human fibroblast nuclei [441]. The required placement locations of adjacent chromosomes are largely independent of the presence or absence of specific neighbors since the positions of CTs relative to each other is highly variable [442, 443], though the tandem arrays of rDNA genes on chromosomes #13, #14, #15, #21 and #22 constituting the nucleolar-organizing regions should be clustered [362]. Minor errors in chromatin positioning by the Proboscis should not crucially affect the outcome because (a) interphase chromosomes are naturally present over a fairly wide range of nuclear radii [441], (b) such chromosomes may be significantly intermingled when decondensed [444], and (c) the precise organization of chromatin into domains varies from mother to daughter cell [445]. Spontaneous chromosome folding during interphase can be inferred by computational analysis of chromosome sequence, using the gene density profile as a manifestation of the “folding code” [446]. Protein or other factors that might be necessary to elicit complete CT self-assembly (possibly including matrix components [447], mechanoenzymes or motor proteins), including histone-chromatin associations [448] and other higher-order chromatin structures [449], can be released through the Proboscis during chromatin placement as needed. The morphology and contents of accessory intranuclear compartments containing such factors are highly dynamic [362]. CTs appear to be probabilistically assigned [441, 442] and systematic maps have been prepared [441]. The radial position of a given CT (or segments thereof) is correlated with its size and gene density [450], its transcriptional activity [451], and its replication timing [450], and also with cytotype [442, 443] since most cell types only express a relatively small fraction of their genome and because genes can be silenced if positioned into regions of chromosomes that are not accessible to chromatin remodeling factors or transcriptional activators [452]. Specifically, small gene-rich and early-replicating chromosomes (e.g., #17, #19, #20) concentrate toward the center of the nucleus, typically without any apparent connection to the nuclear envelope [453], whereas small gene-poor [454] and later-replicating chromosomes (e.g., #18, Y) aggregate toward the nuclear periphery and are attached at the nuclear envelope [450, 453, 455], an evolutionarily conserved organization from vertebrates [450] through primates [456] to humans [453]. The transcriptionally inactive X chromosome localizes peripherally in female cells [451]. CT size is a somewhat larger influence than gene density [457], with small chromosomes distributed significantly closer to the center of the nucleus and large chromosomes preferentially located towards the nuclear periphery [441, 453]. (In contrast to these highly reproducible radial arrangements, 2D distances measured between heterochromatin blocks of homologous and heterologous CTs are quite variable [453].) The head-to-tail orientation of the chromatin strand during placement relative to nuclear center or periphery is also important – e.g., centromeric regions of at least ten chromosomes are localized on the nuclear periphery, often making contact with the nuclear membrane, while telomeric domains are consistently localized within the interior 50% of the nuclear volume [458] – and may be controlled by the packing direction of the chromosome, inside the nanorobot, which will be preserved during viscous extrusion. Chromosome loci typically have confinement radii <0.5 micron [201], so relatively little post-placement movement of chromosomes and subchromosomal domains is anticipated [459]. (18) Decondense chromatin. The condensed chromatin must be decondensed, distributing throughout the nucleus [459] in an unpacked form that will allow access to genes by the transcription apparatus. The chromallocyte can elicit decondensation by releasing an engineered synthetic factor analogous in action to trichostatin A (TSA)-induced histone acetylation [460], nucleoplasmin [461], mimosine [462], or prothymosin alpha (ProTalpha) [463], or by manipulating ion concentrations [32]. Natural mitotic decondensation takes from ~0.5 hr for compact chromatin domains up to ~2 hours for less compact chromatin and nucleoli [459], but chemically induced decondensation of chromatin prepositioned in proper territories should proceed much faster since even natural decondensation motions can apparently occur as fast as ~1 micron/min [459]. The precise mechanism of natural decondensation is presently unknown but likely involves the action of chromokinesin mechanoenzymes [464] such as the human chromokinesin KIF4 which is predominantly associated with the nuclear matrix and has a hexapeptide PKLRRR (amino acids 773-778) that functions as a nuclear localization signal, suggesting that human KIF4 might be a mitotic nuclear motor molecule with DNA as its cargo [465]. The nucleus also contains structural actin, actin-binding proteins, and myosin motor molecule isoforms [466], and NuMA [467] has the dynamic capacity to form lattices [468]. Other motor molecules may rotate chromatin within CTs [469]. (19) Re-anchor chromatin. Upon decondensation, chromatin must be re-anchored to lamin B attachment points in the INM. Integral INM proteins LAP2 and LBR mediate the reassociation of nuclear membranes with chromosomes at the end of mitosis [470]. Since phosphorylation of LAP2 by mitotic cytosol inhibits its binding to both lamin B1 and chromosomes [471], such binding could be enabled by dephosphorylation of LAP2 using a LAP2-targeted engineered synthetic protein phosphatase analogous to those known to dephosphorylate lamin B [472] or histones [473], that could be released, then later reacquired, by the chromallocyte. (20) Reactivate transcription. DNA transcription activities in the nucleus (producing mRNA for export to cytoplasm) must be resumed. Reversible transcription inhibitors that were released in Step 11 are now reacquired by the chromallocyte, using sorting rotors possessing binding sites for the inhibitors. The acquisition rate for 10 kD molecules at 1 nanomolar concentration in nucleoplasm (~100 molecules) using 1000 sorting rotors is initially ~10 molecule/sec [39rr], falling to ~0.1 molecule/sec at 0.01 nanomolar (i.e., the last molecule to be removed), hence nucleosol clearance time is ~500 sec. This pickup rate is consistent with the known <100 sec transnuclear diffusion time of inhibitor proteins and protein complexes up to ~500 kD [362]. A bank of 1000 rotors each having 20 times the volume of the ~2 nm radius 10 kD molecule target occupies <0.001 micron3 of nanorobot hull volume. Cajal bodies or CBs [474], SFCs [475] and the perichromosomal sheath [476] are mitotic repositories for proteins required for a variety of interphase cellular processes, including the synthesis of messenger RNA, assembly of ribosomes, repair of DNA double-strand breaks, telomere maintenance, and apoptosis regulation. Association of proteins in the perichromosomal layer [476] occurs from prophase until telophase, and CBs also preferentially localize some of these proteins [477]. A similar intranuclear penumbra of protein factors should evade the mechanical chromatin detachment/spooling process of CRT that occurs nearer the nuclear center. These factors should remain available for subsequent transcriptional activities because turnover of nuclear components is minimal [477a] and because rapid exchange of proteins has been observed between the nucleoplasm and virtually all nuclear compartments including the nucleolus, SFCs, CBs, PNCs, and heat-shock granules [362]. For example, more than 10,000 molecules of the rRNA processing enzyme fibrillarin are released each second from the 2-3 nucleoli in a cell nucleus [93]. The linker histone H1, which resides outside of nucleosomes but acts as a structural chromatin protein, is dynamically exchanged between binding sites [478], with each molecule of H1 residing for only ~1 minute on chromatin before it dissociates [91]. Another group of structural chromatin-binding proteins, the high-mobility group proteins, associate only transiently with chromatin, residing on chromatin for less than 5 seconds [93]. Nucleologenesis [405] is concurrent with the reinitiation of transcription following resumption of RNA polymerase I function [479] and re-localization of some soluble factors (UBF, nucleolin, fibrillarin) [480]. Introduction of rDNA minigenes results in the formation of mini-nucleoli in the nucleoplasm with nucleolar components accumulating at rDNA-transcription sites [362], directly demonstrating that resumption of rDNA transcription should be sufficient for the assembly of the nucleolus [481, 482]. DNA transcription now resumes after a total hiatus of ~4700 sec (1.3 hr). During the hiatus, protein synthesis has proceeded as normal in the cytoplasmic compartment because typically about half of the mRNA in mammalian tissue cells has a ~6 hour half-life against cytosolic RNAase degradation and the other half of the mRNA has a ~24 hour half-life (~cell cycle length) [87], though differentiated cells devoted to synthesis of specific products have even greater stability and a few eukaryotic mRNAs are less stable. Since only (1-(1/2)(1.3)/6) ~ 14% of cytosolic mRNA may naturally degrade during the transcription hiatus, most metabolic and regulatory cell functions should remain normal during CRT. If mRNA degradation becomes a serious concern, the stability of some mRNAs may be regulated by external agents such as hormones [87] whose concentration may be manipulated by the chromallocyte. Alternatively, cytosolic RNAases could be temporarily extracted from the cytosol using molecule-specific pumps on the surface of the chromallocyte or the nanorobot could release (then later reacquire) reversible RNAase inhibitors, thus extending the active lifespan of existing mRNA strands already present in the cytoplasm, during CRT. (21) Remove nuclear blockades. Engineered inflammation signal inhibitors or enzymes released during Step 10 are reacquired by the chromallocyte, and blockage of DNA repair (Step 9) and apoptosis (Step 8) pathways is reversed by extraction of relevant factors using molecular sorting rotors. The Proboscis is fully retracted to its original interior position. (22) Reverse nucleopenetration. Retracing Step 7, the chromallocyte slowly withdraws from the nucleus, allowing the breached surface of the nuclear membrane to self-seal [39jj, 40i], as commonly occurs, evidently without mishap, during pronuclear microinjection [33]. Simple gene mutations have elicited a nuclear envelope seal that covers nuclear pore openings [483] or nuclear envelope herniations that extend into the cytoplasm [484], and the ER can provide new material to fill holes in mechanically damaged nuclear membrane [485]. Nuclear membrane breaches also might be induced to re-seal by the nanorobot release of an engineered synthetic GTPase similar to Ran [486], since smoothed nuclear envelopes incorporating nuclear pores have been assembled around Ran-coated beads, forming double-walled pseudo-nuclei that actively imported nuclear proteins even in the absence of chromatin. Just as two haploid nuclei held in close proximity will rapidly fuse to form a single diploid nucleus during karyogamy [487], similarly a flap of vacated nuclear envelope wall should spontaneously fuse with the larger section from which it was cut. The chromallocyte might accelerate this process by releasing along the seam of the fissure an engineered synthetic nuclear envelope fusogen similar in function to (a) the BiP-activated ER-localized fusogen that elicits nuclear fusion and karyogamy in yeast [488], (b) phosphatidylinositol-induced diacylglycerol that elicits fusion of nuclear envelope membrane precursor vesicles following mitosis [489], or (c) the EFF-1 fusogen protein for cell-cell fusion [490], red cell fusogen [491], the mitochondrial fusogen protein ORF640p [492], or the tetraspanin fusogen peripherin-2 [493]. (23) Reverse cytopenetration. Retracing Step 4, the nanorobot withdraws from the cell, passing through the cell plasma membrane out into the extracellular regions. Plasma membrane is readily resealed [494] by the chromallocyte [39jj, 40i], possibly employing specialized fusogens [495, 496]. During membrane transit, the nanorobot can pump excess nucleotide-related materials out of the cytosol and into the extracellular region using sorting rotors if necessary in the case of proliferating cells (Section 6.1). Simultaneously, the mechanotransduction blockage initiated in Step 5 should be reversed by retrieving the synthetic mechanotransduction inhibitors previously released into the cell. Diffusion time is ~0.1 sec for 10 kD molecules across ~1 micron paths in cytoplasm [39ab]. (24) ECM emigration. Retracing Step 3, the chromallocytes proceed away from the target cell towards the nearest capillary vessel, through which they originally entered. In some cases, the nanorobot may need to transit 1-2 cells to reach the ECM before arriving at the nearest blood vessel. (25) Reverse transmigration. Retracing Step 2, the nanorobots exit the tissues by undertaking extravasation via basal to apical (reverse) transendothelial migration (aka. reverse transmigration or reverse diapedesis) [497] through the local endothelium to gain entry into the vascular lumina, resealing breaches caused by their passage [39k].
(26) Exit the body. Using guidance from the navigational grid and the previously prepared map, the chromallocytes walk along the blood vessel surface moving away from the target organ until they reach the nanocannula through which they were originally introduced, then exit the body through the nanocannula, the reverse of Step 1. Because the devices do not freely circulate in the blood, they may enter or exit the body through an artery or vein, whichever is most convenient. If a few nanorobots are accidentally left behind, they should still be capable of emergency auto-excretion through the renal tubules as long as external power is still being supplied to the patient, or they can allow themselves to be harmlessly phagocytosed and transported to lymph nodes (Section 6.11). Alternatively, though much less efficiently and with greater risk, the nanorobots could simply pass into the venous system and be removed from the general blood circulation by “chromallocyte affinity filtration” or even nanapheresis [39yy] (e.g., dialysis). 6. Special Cases and Alternate Missions 6.1 Proliferating Cells The standard cell cycle for proliferating cells includes four rigidly-controlled and sequentially-executed phases: entry G1 phase (cell expands in size), S phase (DNA synthesis), G2 phase (resting), and final brief M phase (mitosis or cell division, only ~4% of total cycle duration). Cells not currently replicating are said to be in the quiescent G0 phase; cells may exit M phase into either G0 (to cease proliferating) or G1 (to resume proliferating). In adult organisms, many cell types (e.g., parenchymal cells of the liver and kidney, and glial (brain), retina, muscle (heart), and fibroblast cells) enter the G0 phase semi-permanently and can only be induced to resume dividing under very specific circumstances, while a few other cell types (e.g., bone marrow, epithelial cells, oral/intestinal mucosa, hair follicles) continue to divide throughout an organism’s lifetime. The fraction of proliferating cells in a target organ or tissue varies greatly according to cytotype. For example, the observed growth fraction (GF) (the proportion of actively proliferating cells in G1/S/G2/M phase as a percentage of all viable cells of a given cytotype) is 0.4% for brain glial cells [498], 7.6% for oral mucosa [499], 9.8% for peripheral blood stem cells [500], 10-20% for liver hepatocytes [501], 17.8% for bladder [502], ~40% for skin epidermis [503], and 44% for gut epithelium [504]. G0 cells are readily distinguished from nonquiescent cells by well-known cytochemical markers [505], and other markers can distinguish among the G1 phase (e.g., cyclin D1 [506]), S phase (e.g., cyclin A [506], PCNA [507]), G2 phase (e.g., cyclin B1 [506]), and M phase (e.g., p105 [508], p34(cdc2) [509], mitosin [510]) of a proliferating cell. Chromallocyte activities applicable to quiescent G0 cells (Section 5) must be slightly modified for CRT on nonquiescent cells, as follows. If the cell is found to be in G1 phase during the organ survey (Section 5.1), the survey nanorobot releases into the cytosol an engineered synthetic reversible G1/S phase transition inhibitor perhaps analogous in action to the tumor suppressor retinoblastoma protein pRb that blocks S-phase entry [511], or analogous to cyclin dependent kinase (CDK) inhibitors that suppress cell growth [512], blocking replication licensing and arresting the cell in a G2-like resting state [513]. For example, increased expression of p21waf1/cip1 blocks cdk2 activity in G1 phase, leading to hypophosphorylation of pRb and inhibition of cell cycle progression into S phase [514]. After normal chromosome replacement (Section 5.2) on a cell trapped in G1 phase, the inhibitor is extracted and cell biochemistry is reset to G0 or G1 by directly altering the concentrations of crucial cytochemical factors. Cells found to be in G0 during the survey that subsequently enter G1 cannot have exited G1 phase prior to chromallocyte arrival since the duration of G1 is typically 6-12 hours. If the cell is found to be in S phase or G2 phase, the survey nanorobot releases into the cytosol an engineered synthetic reversible downregulator perhaps analogous to elements of known pathways that mediate rapid arrest in G2 in response to DNA damage by downregulating a large number of genes encoding proteins required for G2 and M, thus eliminating essential cell cycle proteins and keeping the cell arrested at the G2/M checkpoint [515] without triggering apoptosis. A synthetic inhibitor analogous to roscovitine could also be released, non-apoptotically directly inhibiting DNA synthesis in S-phase cells [516]. Following funnel sealing during chromosome replacement (Section 5.2), up to half of the DNA may remain outside the closed funnel (because an entire second genome has been replicated) and thus must be digested in situ (Step 16), using extra nuclease provided onboard for this purpose. Note that whole-genome digestion and extranuclear release would raise cytosolic nucleotide concentration to ~0.002 gm/cm3, 1-3 orders of magnitude higher than normal levels of AMP/GMP in cytoplasm and free nucleotides in blood plasma. Any such excess nucleotide must subsequently be pumped from the cytoplasm to the extracellular space (Step 23) during chromallocyte egress from the cell, at which time the cycle inhibitors also are extracted and cell biochemistry is reset to G0/G1 as required. In the relatively rare case that the cell is found to be in M phase, mitosis will be allowed to run to completion but the survey nanorobot will release into the nucleus-free cytosol an engineered synthetic reversible cell-cycle restart inhibitor analogous in action to the inhibitor protein geminin [517] and other factors [518] that apparently prevent replication licensing by Cdt1, a key component of the origin recognition complex (ORC), thus blocking the cell from reinitiating G1 phase at the normal end of mitosis, without inducing apoptosis [519]. Most eukaryotic cell types can withdraw from proliferative cell cycles and remain quiescent for extended periods [520]. Since two cells will now exit M phase and both will require CRT, the cell map is adjusted accordingly and two chromallocytes instead of one are dispatched to the site. After normal completion of chromosome replacement (Section 5.2), the synthetic restart inhibitor is extracted and cell biochemistry is reset to G0/G1 as required. Cells undergoing mitosis during Phase I survey will complete mitosis in at most ~1 hr, hence should be postmitotic by the start of Phase IV of CRT. 6.2 Pathological Cells Cells with chromatin-related pathologies such as cancer, viral insertions, aging, or genetic disease can be corrected by CRT. Cells with chromatin-unrelated pathologies such as intracellular lipofuscin, crystalloid deposits or other storage diseases, or indwelling viral, bacterial, or parasitic infections should be cleared of these deposits or pathogens [42] before performing CRT. Isolated cells with severe physical or mechanical trauma that are incapable of self-repair might be allowed to enter apoptosis, and any apoptosis in progress should be allowed to continue to completion to provide an orderly alternative to necrosis. However, large numbers of contiguous cells with severe physical trauma (e.g., major wounds) should be surgically replaced or repaired by other nanorobotic instrumentalities prior to CRT. 6.3 Brain, Bone, and Mobile Cells Access to cells for CRT will be more challenging in a few organs. For example, CRT in the brain requires nanorobotic passage through the blood-brain barrier (BBB) [40h], or alternatively through nanocannula placed directly into the neuropil. Chromallocyte mobility systems must carefully avoid mechanical disruption of dendrites and synaptic connections by reducing transit velocities, applied forces, and mechanical frequencies. Mechanical pathologies of neural tissue include spinal cord dysfunction from mechanical compression [521], enhanced vulnerability to secondary insults following sublethal mechanical stretch [522], glial reaction to vestibular nerve dendrite lesions [523], and waves of apoptotic neurodegeneration [524]. In bone, marrow and cancellous bone cells are readily accessible even to large nanorobots due to the open lattice structure [39ss]. However, nanorobots seeking access to osteoblasts and osteocytes located in compact bone must transit out of the 20-micron Haversian canals into smaller fluid-filled canaliculi averaging 0.3 microns in diameter (range 0.1-1.0 micron) [525], necessitating modified nanorobot and mission designs. Dentin cells in the teeth are somewhat more accessible through 1-4 micron diameter dentinal tubules [526, 527], but smaller chromallocytes or multiple cell visits per replacement operation may still be required. For nonfixed nontissue cells such as leukocytes and fibroblasts, cell mobility may preclude a fixed-map-driven CRT mission because, e.g., fibroblasts hasten through ECM at speeds of 200-2000 microns/hr [39g]. However, most mobile cells have short lifetimes – e.g., neutrophil lifespan is ~3 hours in blood, ~3 days in tissue [528], and typically 1-2 months [529, 530] but up to 4-8 months [531, 532] for cultured fibroblasts. These cells will be naturally replaced in due course with new cells that can possess corrected chromosomes, since map-assisted CRT can be performed on the progenitor mesenchymal and hematopoietic source cells [533] that are located at relatively fixed positions in bone marrow and in the lympho-hematopoietic components of other organs (e.g., thymus, spleen, lymph node, Peyer’s patches). If desired, circulating nucleated mobile cells could be extracted from blood via cytapheresis [534], after which these cells may be individually characterized [535] and receive CRT in an ex vivo processing device, then be returned to the patient’s circulation. Similar catch-and-release CRT strategies can be employed for longer-lived tissue-dwelling mobile cells if required. 6.4 Multinucleate Cells A few cell types such as skeletal muscle cells, osteoclasts, megakaryocytes, and some hepatocytes have multiple nuclei of similar size [55]. To perform CRT on multinucleate targets, multiple chromallocytes must be sent, in sequence (to avoid cytoplasmic crowding), to the cell. Upon arrival, the first nanorobot enters the cell, seeks out a nucleus, ascertains that the nucleus has not previously had CRT by verifying the absence of marker (see below), then performs CRT on that nucleus. After exiting the nucleus following CRT and resealing the nuclear envelope (Section 5.2, Step 22), the chromallocyte nanoinjects into the nucleus a small quantity of a low-MW fast-diffusing engineered marker molecule. The marker molecule should be biologically inactive and should possess bulky laminophobic or lipophobic distal end groups to discourage diffusion through the nuclear envelope, but should be capable of degradation into biologically inactive but metabolizable cleavage products by native nuclear enzymes with a half-life conveniently exceeding the anticipated total mission duration (e.g., >24 hours). The second nanorobot to arrive at the cell repeats the search for a nucleus, perhaps finds the marked one and skips it, and proceeds to the next unmarked nucleus where it performs CRT, then again injects marker in the second nucleus to indicate task completion. This procedure continues with successive chromallocytes until all nuclei are tagged with marker. Any surplus nanorobot that arrives subsequently and finds that all available nuclei are marked should exit the cell without performing CRT. Because nanorobots should work sequentially, not concurrently, each additional nucleus in a multinucleate cell population adds ~2 hours to total mission duration (per wave of repair) compared to CRT on mononucleate cell populations. In the case of highly elongated muscle cells containing up to several hundred myonuclei [536], time can be saved by injecting a different binary-coded marker into each nucleus during mapping, with each chromallocyte targeted to a different marker inside that cell. Since myonuclei are well-separated, typically 20-50 microns between nearest-neighbor nuclei [537], the simultaneous intracellular presence of multiple CRT nanorobots should be tolerable. Initial myonuclear locations can be surveyed and mapped as a rough guide for later-arriving chromallocytes that will continue sampling in the vicinity until they detect their encoded target chemical marker. Myonuclei are observed to maintain their relative positions over long periods of time, with internuclear distances typically varying by a mean ~5.9 micron over a 4-day observation period in murine myotubes [537]. 6.5 Karyolobism and Karyomegaly Karyolobism occurs when the cell nucleus is lobed, possibly so deeply lobated or divided that the cell appears to have multiple nuclei. A cell having a single but non-compact nucleus with DNA distributed throughout numerous lobes could be more difficult for a chromallocyte to reliably clear of its chromatin. The most common karyolobate cells are the polymorphonuclear leukocytes (or granulocytes) and include the neutrophils (~60% of all leukocytes), eosinophils (1-4%), and basophils (<1%). All three types are found in the blood but are short-lived and produced from bone marrow progenitors (which are not karyolobate), hence can be replaced (after a time lag) with cells having new DNA by performing CRT on the progenitors and not on the granulocytes themselves (Section 6.3). Karyomegaly is the abnormal enlargement of the cell nucleus, a condition which could make it harder for the chromallocyte, which has limited physical reach, to efficiently spool the old chromatin. Pathological karyomegaly occurs most commonly in kidney cells due to heavy metal [538], chemical [539], and viral [540] nephropathies, but also in liver [541, 542]) and pancreas [542] cells, arterial endothelial cells [543], and in the epithelial and mesenchymal cells of several other organs [544]. For example, mouse thigh muscle nurse cell nuclei of normal mean volume 84 micron3 (range 42-170 micron3) increase 8-fold to a peak mean volume of 681 micron3 (range 407-1138 micron3) by day 8 after infection with Trichinella spiralis [545]. Similar changes also normally occur in proliferating cells, whose nuclei can increase in volume by up to 5-6 times during G1 phase [459], and to a lesser extent in the nuclear volumes of cells of juvenile animals as they mature to adults (e.g., by +79% for rat cardiac myocytes [546]). Note that the 6.2 micron radius of a large 1000 micron3 spherical nucleus is significantly exceeded by the maximum (LProb + Zext ) ~ 9.05 micron reach of a nuclear-envelope-anchored nuclear-indwelling chromallocyte. Mitochondriomegaly – an abnormal swelling by up to +236% of DNA-containing mitochondrial organelles during disease – has also been observed [547, 548], with similar relevance to prospects for mitochondrial CRT (Section 6.6). 6.6 Mitochondrial DNA Mitochondria are scattered throughout the cytoplasm in virtually all aerobic cells, with numbers ranging from ~300/cell for relatively inactive cells like lymphocytes up to 2000-3000/cell for very active cells such as liver, kidney tubule and cardiac muscle cells (where mitochondria may occupy up to 20% of total cell volume [70]). Uniquely in animal cells outside of the nucleus, mitochondria contain their own DNA (mtDNA) though this role as a genetic vault may be incompatible with the role of these organelles in bioenergetics since electron transport results in the generation of reactive oxygen species that can sometimes induce lesions in the mtDNA [549]. This “design error” is possibly remediable by constitutively offloading transgenic copies of mtDNA onto the nuclear chromosomes (proposed for treating disease [550] and aging [551, 552] and first partially demonstrated experimentally in 1986 [553]), an interesting subject that nevertheless lies beyond the scope of this paper. Many classically untreatable mitochondrial diseases that arise from single homoplasmic mutations in mtDNA typically affect nonmitotic brain, retina, or muscle tissues. Evolving evidence also implicates mtDNA abnormalities in diseases such as Alzheimer’s, Parkinson’s, and type II diabetes though specific causal mutations for these conditions remain imprecisely defined [554]. These problems have led to proposals for mitochondrial gene replacement therapy using the traditional techniques of biotechnology [552, 555], possibly using engineered organisms perhaps derived from known bacterial symbionts than can live inside mitochondria [555a]. Chromallocytes may offer a more efficient approach. CRT is normally performed only on nuclear chromatin, but mtDNA could be replaced using chromallocyte-class devices and a revised treatment protocol. The typical ~1 micron human mitochondrion possesses a genome [556] that includes 5-10 identical double-stranded circular DNA molecules each of ~5 micron contour length encoding 22 tRNA genes, 2 rRNA genes, and 13 protein-coding regions using ~16,569 base pairs [87]. Even for cells in which 3000 mitochondria are present, the total DNA to be replaced would be at most ~500 million base pairs per cell or ~8% of the normal chromallocyte payload, allowing mitochondrial-targeted chromallocytes to be much smaller in size but requiring redesigned Proboscis and funnel assemblies for efficient mtDNA extraction and replacement. The replacement procedure would be similar to the marker-based process described in Section 6.4 for multinucleate cells, except that all mitochondrial organelles within a target cell would have their DNA replaced by a single nanorobot during one visit. Since a purely random catch-and-release strategy would produce unacceptably long search times once most, but not all, mitochondria had been processed, demobilization and positional sequestration of the organelles as they are processed would greatly reduce total CRT time and better ensure task completion. Alternatively, a possibly more efficient negative-pass system could be employed in which a hypothetical engineered marker molecule, capable of localizing only to the interior fluid of mitochondria [557] and also capable of targeting mitochondria for lysosomal digestion [558] (aka. “mitoptosis” [356] or “mitophagy” [559]) once naturally cleaved inside the organelle after a lengthy degradation time delay, is diffused into the cytosol by the nanorobot, then extracted from the cytosol after an appropriate waiting time to ensure mitochondrial uptake. Marker would then be extracted from any organelle receiving CRT from the nanorobot, thus ultimately preserving it, but leaving unmodified mitochondria to their marker-induced digestive fate [560, 561]. Any resulting shortage of mitochondria in the cell would be rapidly eliminated by replication of surviving mitochondria [562] upon which CRT had been performed. It might also be useful to chemically induce mitochondria to eject their mtDNA into the cytosol [563, 564], thus permitting convenient enzymatic degradation prior to CRT, if new methods can be found that avoid destroying the organelle. 6.7 Nonpathological Mosaicism Genetic mosaicism is the presence of two distinct genomes in the cells of the same individual, as in nonpathological X-chromosome inactivation (Section 4.1) which is a form of non-DNA-sequence mosaicism, or as in various pathological conditions related to somatic mosaicism [565], germ line (gonadal) mosaicism [566], or mitochondrial heteroplasmy [567], which the patient might wish to correct (Section 4.1). The most important example of nonpathological mosaicism is found in the immune system where B cells each display just one of ~108-109 possible antigen receptor types, with ~107 different specificities occurring in a single individual [568]. Since the amino acid sequence comprising the receptor protein is encoded in B-cell DNA, there are ~107 unique genomes that may be found in this cell population within each patient, with each genome differing very slightly from the others primarily by the coding of 30-60 amino acids in the hypervariable or V(D)J recombination domains containing most of the epitope-specific sequence variation [569]. B cells are concentrated in lymph and lymph nodes. When activated by antigen, B cells replicate and differentiate into large number of antibody-producing plasma cells with a ~1 week lifetime, then a few plasma cells differentiate into memory B cells that are stored in the germinal centers of lymphoid organs to maintain a long-term record of past antigens encountered. Performing information-preservative CRT on the immune system would require sampling and sequencing all ~107 unique specificities of the individual patient (directly sampled from genomes taken from the ~1012-cell B cell and T cell lymphocyte populations or inferred by sampling the ~1020 circulating free immunoglobulin molecules), then replacing the genomes of the relatively fixed stem cell and germinative cell populations with an appropriate number of randomly distributed new genomes encoding all ~107 specificities, thus preserving the patient’s original mix of antigen reactivity. A catch-and-release cytapheresis-based CRT process (Section 6.3) could also be applied to circulating nucleated B cells and T cells in an ex vivo processing device, if desired. Alternatively, patients could be provided with a more robust set of antigenic reactivities as a supplement or replacement to their own, possibly dramatically improving immune response. Immunoglobulin genes [570] and the related T cell receptor (TCR) genes [569] may constitute the largest gene family, representing >2% of the human genome and residing on multiple chromosomes, but the total amount of hypervariable information to be extracted is probably less than one short chromosome. A ~3 cm3 block of nanomechanical sequencing hardware (Section 4.2) could sequence ~104 chromosome-equivalents/sec, thus processing 100 copies x 107 specificities in ~105 sec or about 1 day. All else equal, chromatin manufacturing and chromallocyte operational times should be largely unchanged. Limited mosaicism may also take place during brain development due to retrotransposition events in LINE-1 elements that can alter the expression of neuronal genes and produce mosaicism in adult neurons derived from a single original neuronal genome in that patient [571]. If this process is confirmed, similar CRT strategies can be employed and refined by future work.
6.8 Partial- or Single-Chromosome CRT Early proposals for cell repair nanorobots [572] envisioned intracellular in situ diagnosis and repair on a molecule-by-molecule basis. But replacing a single defective nucleotide or a single defective gene on a given chromosome, rather than replacing the entire chromosome, would require controlled spooling to the exact site needing repair. It might risk possible inadvertent activation of nearby cancer-causing genes, or other unwanted genes, analogous to retroviral oncogenesis [573], if new material, added in situ, is improperly spliced in. Similarly, identifying, extracting, and replacing just one chromosome from among many in the nucleus might demand chemically-modifying and relatively time-consuming in situ chromosome identification processes analogous to FISH [574], might require additional chromatin sequencing and manufacturing facilities to be built into the nanorobot, will require the same procedural overhead (e.g., implementing reversible apoptosis blocking, etc.), and would risk disruption of existing territories and mechanical damage to adjacent chromosomes which are not being replaced. The whole-genome replacement strategy also is affirmed by the evolved natural process of highly reliable cell replication, in which a cell replicates just once every DNA sequence in the nucleus, and having initiated replication must pursue it to completion with a whole genome. Biology never tries to replicate just one chromosome at a time, possible evolutionary evidence that piecemeal replacement may invite unacceptable error. The whole-genome strategy also permits all defects on multiple chromosomes to be efficiently corrected at once, avoiding subsequent repeat visits to the same cell to correct other deficiencies. At the other extreme, replacing existing whole nuclei with premanufactured nuclei using mobile nanorobots seems difficult because the typical ~268 micron3 nuclear volume [39a], even if compacted by partial dehydration [40r], would require a 200-300 micron3 delivery vehicle, too large to conveniently traverse the capillaries. A non-nanorobot pipette-based strategy for performing nuclear replacement on trillions of closely-packed cells in otherwise undisturbed organs seems unduly intrusive but cannot be ruled out. However, the multiple shapes (Section 6.4) and sizes (Section 6.5) of nuclei complicate the mission. Mechanical enucleation also would require sequestration of a much larger quantity of waste material, would require a more aggressive suppression of mechanotransduction and apoptotic responses throughout the cell than for CRT, and would severely disrupt the existing ER apparatus and the numerous cytoskeletal associations with the nucleus.
6.9 Single-Cell and Whole-Body CRT Chromallocytes might be targeted to perform CRT on sub-organ-sized groupings of cells such as (a) fixed-tissue micrometastases [575] or migrating [576] tumor cell clusters (but not to circulating cancer cell clusters [577, 578] which should be destroyed, not repaired), (b) localized bone joint senescent chondrocytes [579], (c) hair follicle cells in balding scalp [580], and so forth. Targeting small numbers of isolated individual cells for CRT is feasible but seems inefficient. At the other extreme, whole-body CRT is feasible but the process would take considerably longer than 7 hours. As a crude estimate, if chromosome replacement can be performed on ~1012 tissue cells in 4 hours (Section 5.1), then extending the procedure to all ~1013 tissue cells in the body [39tt] implies a ~43 hour treatment time for continuously-performed whole-body CRT, with special cases possibly adding another 5-10 hours to the total. This estimate assumes that survey operations in one organ can proceed in parallel with chromatin replacement operations in another previously surveyed organ. However, all cells cannot be processed at once because the simultaneous operation of >1013 chromallocytes at a 50-200 pW power draw would dissipate >1000 watts inside the human body, well in excess of the ~100 watt maximum safe thermogenic limit in vivo [39ac].
6.10 Heteroiatrogeny One potential occasional risk inherent in single-organ CRT is “heteroiatrogeny” – a potentially adverse state caused by medical treatment of some organs but not others, informally known as “car repair syndrome.” Just as replacing one broken part in an aging automobile with a new part can put sudden fatal extra stress on the remaining older original parts, restoring just a single organ to youthful levels of performance might put stress on the remaining unrepaired organs, hastening their failure as well. For example, restoring only the hormonal organs of a geriatric patient to full youthful vigor might encourage the patient to exercise more violently than his other aging organs could tolerate, possibly leading to torn ligaments and muscle damage [581], bone fractures [582] and osteoporosis [583], organ changes [584], other symptoms analogous to overtraining [585], and other serious injuries such as heart failure or stroke. Similarly, if a single repaired organ of a pair (e.g., one of the two lungs or kidneys) functions better alone than had both of the original aging organs working together, this could remove stress from the unrepaired organ and allow it to atrophy. The risks in heteroiatrogeny thus may argue in favor of whole-body CRT, a modern redux of holistic medicine [586] in the nanomedical era.
6.11 Nanorobot Malfunction Nanorobots are designed with tenfold redundancy of most mission-critical components, double the lesser five-fold redundancy of the Space Shuttle computers [587]. Systematic failure rates for nanomachines are dominated by radiation damage effects. Adopting Drexler’s [51c] nanomachine radiation damage model and applying it to a system comprised of Nc components, with each component comprised of n redundant parts, the probability Pop that the system remains operational after Tmission years is approximated by: Pop = exp [-Nc (1 - pop)n], where pop = the probability that an individual part remains operational; pop ~ exp(-1015 Dradmpart), where mpart = mass of the part in kg and Drad = radiation dose in rads ~ 0.3 Tmission for normal background radiation in the terrestrial environment. Assuming that all chromallocyte modular “parts” are similar in mass (mpart ~ 10-19 kg) to the ~4,000,000-atom nanorobotic manipulator arm described elsewhere [51d], then the ~4 trillion-atom chromallocyte has at most Nc ~ 106 such components each with pop ~ 0.99997 for Tmission = 1 year. Even using only fivefold redundancy (n = 5), Pop ~ 1 - 10-16 which implies a failure rate of ~1 nanorobot per year per 10,000 trillion nanorobots, or a ~0.1% probability of a single nanorobot failure per whole-body CRT procedure. With tenfold redundancy, intrinsic failure rates are even lower. Other modes of nanorobot malfunction such as nonredundant equipment failure (e.g., jammed Proboscis, vault leak, etc.), engulfment by phagocytes [40k], biofouling [40m], cross-interface adhesions [40n], computer failure [39uu] or navigational errors [39vv] seem more likely but are also expected to occur very infrequently. In the rare event that a chromallocyte becomes immobilized in cyto during a replacement operation, the affected transcription-deactivated cell will soon exhaust its remaining mRNA and suffer a necrotic death, eventually releasing the inert nanorobot into the surrounding intercellular medium. In this case, or in the case of a nanorobot that fails before reaching its cellular target, the inert nanorobot ultimately will be taken up by phagocytes which may then be harmlessly granulomatized at the site for indefinite periods [40o, 40p] or may be swept into the lymphatic circulation resulting in permanent harmless sequestration in the lymph nodes [40q]. If desired, the patient may be post-operatively resurveyed and all such accumulations can be removed by scavenger nanorobots specialized for this purpose.
7. Conclusions This paper reports the first theoretical scaling analysis and mission design for a cell repair nanorobot. The chromallocyte is a hypothetical mobile cell-repair nanorobot whose primary purpose is to perform chromosome replacement therapy (CRT), in which the entire chromatin content of the nucleus in a living cell is extracted and promptly replaced with a new set of prefabricated chromosomes which have been artificially manufactured as defect-free copies of the originals. The chromallocyte will be capable of limited vascular surface travel into the capillary bed of the targeted tissue or organ, followed by extravasation, histonatation, cytopenetration, and complete chromatin replacement in the nucleus of the target cell, and ending with a return to the bloodstream and subsequent extraction of the device from the body at the original infusion site, completing the CRT mission. Replacement chromosomes are manufactured in a desktop ex vivo chromosome sequencing and manufacturing facility, then loaded into the nanorobots for delivery to specific targeted cells during CRT. The CRT mission involves a 5-phase procedure, one of which includes a specific 26-step process for performing whole-genome chromosome exchange nanosurgery on a living cell, using the chromallocytes. A single lozenge-shaped 69 micron3 chromallocyte measures 4.18 microns and 3.28 microns along cross-sectional diameters and 5.05 microns in length, typically consuming 50-200 pW in normal operation and a maximum of 1000 pW in bursts during outmessaging, the most energy-intensive task. Treatment of an entire large human organ such as a liver, involving simultaneous CRT on all 250 billion multinucleate hepatic tissue cells, might require the localized infusion of a ~1 terabot ~69 cm3 chromallocyte dose in a 1-liter 7% saline suspension during a ~7 hour course of therapy. Modified procedures are potentially available for special cases including (1) proliferating, pathological, multinucleate, and karyolobate cells, (2) cells in locations where access is difficult such as brain, bone, or mobile cells, and (3) cells expressing genetic mosaicism, and also for alternative missions including (1) partial- or single-chromosome replacement, (2) single-cell and whole-body CRT, and (3) mitochondrial DNA replacement.
Acknowledgments The author thanks Robert Bradbury, Greg Fahy, Ralph Merkle, Chris Phoenix, Tihamer Toth-Fejel, Brian Wowk, and several unidentified referees for helpful comments and discussions, Forrest Bishop for use of the microbivore grapple images, and John Luu and his colleagues at Stimulacra LLC for creating the 3D models of the chromallocyte that appear in this paper. The author acknowledges research grants from Alcor Foundation, Life Extension Foundation, Kurzweil Foundation, and the Institute for Molecular Manufacturing in support of this work.
References 1. M. Fenech, “Chromosomal damage rate, aging, and diet,” Ann. N.Y. Acad. Sci. 854(November 1998):23-36. 2. M.R. Jones, K. Ravid, “Vascular smooth muscle polyploidization as a biomarker for aging and its impact on differential gene expression,” J. Biol. Chem. 279(13 February 2004):5306-5313. 3. Curtis A. Machida, ed., Viral Vectors for Gene Therapy: Methods and Protocols, Humana Press, 2002.
4. N. Loewen, E.M. Poeschla, “Lentiviral vectors,” Adv. Biochem. Eng. Biotechnol. 99(2005):169-191.
5. S.S Ghosh, P. Gopinath, A. Ramesh, “Adenoviral vectors: a promising tool for gene therapy,” Appl. Biochem. Biotechnol. 133(April 2006):9-29. 6. D. Larocca, M.A. Burg, K. Jensen-Pergakes, E.P. Ravey, A.M. Gonzalez, A. Baird, “Evolving phage vectors for cell targeted gene delivery,” Curr. Pharm. Biotechnol. 3(March 2002):45-57. 7. G. Vassaux, J. Nitcheu, S. Jezzard, N.R. Lemoine, “Bacterial gene therapy strategies,” J. Pathol. 208(January 2006):290-298. 8. H. Yamada, A. Kunisato, M. Kawahara, C.G. Tahimic, X. Ren, H. Ueda, T. Nagamune, M. Katoh, T. Inoue, M. Nishikawa, M. Oshimura, “Exogenous gene expression and growth regulation of hematopoietic cells via a novel human artificial chromosome,” J. Hum. Genet. 51(2006):147-150. 9. S. Chen, J.H. Ding, R. Bekeredjian, B.Z. Yang, R.V. Shohet, S.A. Johnston, H.E. Hohmeier, C.B. Newgard, P.A. Grayburn, “Efficient gene delivery to pancreatic islets with ultrasonic microbubble destruction technology,” Proc. Natl. Acad. Sci. (USA) 103(30 May 2006):8469-8474. 10. C.R. Safinya, K. Ewert, A. Ahmad, H.M. Evans, U. Raviv, D.J. Needleman, A.J. Lin, N.L. Slack, C. George, C.E. Samuel, “Cationic liposome-DNA complexes: from liquid crystal science to gene delivery applications,” Philos. Transact. A Math. Phys. Eng. Sci. 364(15 October 2006):2573-2596. 11. C. Dufes, I.F. Uchegbu, A.G. Schatzlein, “Dendrimers in gene delivery,” Adv. Drug Deliv. Rev. 57(14 December 2005):2177-2202. 12. F.D. Urnov, J.C. Miller, Y.L. Lee, C.M. Beausejour, J.M. Rock, S. Augustus, A.C. Jamieson, M.H. Porteus, P.D. Gregory, M.C. Holmes, “Highly efficient endogenous human gene correction using designed zinc-finger nucleases,” Nature 435(2 June 2005):646-651. 13. S.L. Vanderbyl, B. Sullenbarger, N. White, C.F. Perez, G.N. MacDonald, T. Stodola, B.A. Bunnell, H.C. Ledebur Jr., L.C. Lasky, “Transgene expression after stable transfer of a mammalian artificial chromosome into human hematopoietic cells,” Exp. Hematol. 33(December 2005):1470-1476. 14. M.W. Konstan, P.B. Davis, J.S. Wagener, K.A. Hilliard, R.C. Stern, L.J. Milgram, T.H. Kowalczyk, S.L. Hyatt, T.L. Fink, C.R. Gedeon, S.M Oette, J.M. Payne, O. Muhammad, A.G. Ziady, R.C Moen, M.J. Cooper, “Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution,” Hum. Gene Ther. 15(December 2004):1255-1269. 15. D.J. Bharali, I. Klejbor, E.K. Stachowiak, P. Dutta, I. Roy, N. Kaur, E.J. Bergey, P.N. Prasad, M.K. Stachowiak, “Organically modified silica nanoparticles: a nonviral vector for in vivo gene delivery and expression in the brain,” Proc. Natl. Acad. Sci. (USA) 102(9 August 2005):11539-11544. 16. H.C. Ertl, “Challenges of immune responses in gene replacement therapy,” IDrugs 8(September 2005):736-738. 17. N.I. Wolf, E.A. Sistermans, M. Cundall, G.M. Hobson, A.P. Davis-Williams, R. Palmer, P. Stubbs, S. Davies, M. Endziniene, Y. Wu, W.K. Chong, S. Malcolm, R. Surtees, J.Y. Garbern, K.J. Woodward, “Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus-Merzbacher disease,” Brain 128(April 2005):743-751. 18. D.P. O’Malley, C. Poulos, M. Czader, W.G. Sanger, A. Orazi, “Intraocular inflammatory myofibroblastic tumor with ALK overexpression,” Arch. Pathol. Lab. Med. 128(January 2004):e5-7. 19. R. Gerlai, J. Roder, “Abnormal exploratory behavior in transgenic mice carrying multiple copies of the human gene for S100 beta,” J. Psychiatry Neurosci. 20(March 1995):105-112. 20. J.R. Lupski, “Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits,” Trends Genet. 14(October 1998):417-422. 21. A. Dufke, S. Singer, S. Borell-Kost, M. Stotter, D.A. Pflumm, U.A. Mau-Holzmann, H. Starke, K. Mrasek, H. Enders, “De novo structural chromosomal imbalances: molecular cytogenetic characterization of partial trisomies,” Cytogenet. Genome Res. 114(2006):342-350. 22. M. Lessard, A. Herry, C. Berthou, M.C. Leglise, J.F. Abgrall, I. Luquet, N. Dastugue, E. Duchayne, F. Rigal-Huguet, M. Lafage, D. Sainty, J. Reiffers, P. Bernard, “Chromosome 8 tetrasomies and pentasomies – a clonal abnormality closely associated with acute monocytic leukaemia,” Leuk. Lymphoma 27(September 1997):127-135. 23. O. Fernandez-Capetillo, A. Nussenzweig, “Aging counts on chromosomes,” Nat. Genet. 36(July 2004):672-674. 24. V. Wacheck, U. Zangemeister-Wittke, “Antisense molecules for targeted cancer therapy,” Crit. Rev. Oncol. Hematol. 59(July 2006):65-73. 25. S. Babiuk, S. van Drunen Littel-van den Hurk, L.A. Babiuk, “Delivery of DNA vaccines using electroporation,” Methods Mol. Med. 127(2006):73-82. 26. H. Aslan, Y. Zilberman, V. Arbeli, D. Sheyn, Y. Matan, M. Liebergall, J.Z. Li, G.A. Helm, D. Gazit, Z. Gazit, “Nucleofection-based ex vivo nonviral gene delivery to human stem cells as a platform for tissue regeneration,” Tissue Eng. 12(April 2006):877-889. 27. U.K. Tirlapur, K. Konig, “Targeted transfection by femtosecond laser,” Nature 418(2002):290-291; http://www.nature.com/nsu/020715/020715-7.html 28. A. Melman, N. Bar-Chama, A. McCullough, K. Davies, G. Christ., “hMaxi-K gene transfer in males with erectile dysfunction: results of the first human trial,” Hum. Gene Ther. 17(30 November 2006). 29. B.R. Korf, E.G. Diacumakos, “Microsurgically-extracted metaphase chromosomes of the Indian muntjac examined with phase contrast and scanning electron microscopy,” Exp. Cell Res. 111(January 1978):83-93. 30. B.R. Korf, E.G. Diacumakos, “Absence of true interchromosomal connectives in microsurgically isolated chromosomes,” Exp. Cell Res. 130(December 1980):377-385. 31. M. Frey, T. Koller, M. Lezzi, “Isolation of DNA from single microsurgically excised bands of polytene chromosomes of Chironomus,” Chromosoma 84(1982):493-503. 32. A.J. Maniotis, K. Bojanowski, D.E. Ingber, “Mechanical continuity and reversible chromosome disassembly within intact genomes removed from living cells,” J. Cell Biochem. 65(April 1997):114-130. 33. R.J. Wall, “Pronuclear microinjection,” Cloning Stem Cells 3(2001):209-220. 34. N. Shimizu, F. Kamezaki, S. Shigematsu, “Tracking of microinjected DNA in live cells reveals the intracellular behavior and elimination of extrachromosomal genetic material,” Nucleic Acids Res. 33(3 November 2005):6296-6307. 35. E. Wolf, W. Schernthaner, V. Zakhartchenko, K. Prelle, M. Stojkovic, G. Brem, “Transgenic technology in farm animals – progress and perspectives,” Exp. Physiol. 85(November 2000):615-625. 36. A. Meissner, R. Jaenisch, “Mammalian nuclear transfer,” Dev. Dyn. 235(September 2006):2460-2469. 37. J.B. Gurdon, “Nuclear transplantation in Xenopus,” Methods Mol. Biol. 325(2006):1-9. 38. T. Tada, “Nuclear reprogramming: an overview,” Methods Mol. Biol. 348(2006):227-236. 38a. A. Heddi, A.M. Grenier, C. Khatchadourian, H. Charles, P. Nardon, “Four intracellular genomes direct weevil biology: Nuclear, mitochondrial, principal endosymbiont, and Wolbachia,” Proc. Natl. Acad. Sci. (USA) 96(8 June 1999):6814-6819. 39. R.A. Freitas Jr., Nanomedicine, Volume I: Basic Capabilities, Landes Bioscience, Georgetown, TX, 1999; http://www.nanomedicine.com/NMI.htm; (a) Table 8.17, (b) 5.2.1, (c) 8.2.1.2, (d) 6.5.6C, (e) 9.3.1.4, (f) 5.3.6, (g) 9.4.4.2, (h) 9.4.2.4, (i) Table 9.4, (j) 8.5.3.2, (k) 9.4.4.1, (m) Figure 9.23, (n) 9.4.2.5.1, (p) Table 9.3, (q) 10.3.1, (r) 10.4.2.5.2, (s) 3.4.2, (t) 6.4.1, (u) 6.2.2.2, (v) 6.4.3.4, (w) 8.3.3, (x) 8.3.5, (y) 7.2.1, (z) 10.4.2.4.2, (aa) 9.3.2, (bb) 9.3.1, (cc) 9.4.4, (dd) 3.5, (ee) 9.4.3, (ff) 8.5.3.11, (gg) 8.5.4.6, (hh) 9.4.4.3, (ii) 9.4.5, (jj) 9.4.5.6, (kk) 9.4.5.3, (mm) 9.4.5.7, (nn) Table 8.1, (oo) 9.4.5.5, (pp) Chapter 4, (qq) 9.4.6, (rr) 4.2.3, (ss) 8.2.4, (tt) 8.5.1, (uu) 10.2, (vv) 8.3, (ww) 9.4.3.5, (xx) 9.4.2.5.4, (yy) 10.3.6, (zz) Table 3.2, (ab) 8.5.3.3, (ac) 6.5.2. 40. R.A. Freitas Jr., Nanomedicine, Volume IIA: Biocompatibility, Landes Bioscience, Georgetown, TX, 2003; http://www.nanomedicine.com/NMIIA.htm; (a) 15.4.2, (b) 15.2.2.4, (c) 15.6.3, (d) 15.5.2.3, (e) 15.5.2.2, (f) 15.5.4, (g) 15.5.4.1, (h) 15.3.6.5, (i) 15.5.7.2.2, (j) 15.5.3.1.2, (k) 15.4.3, (m) 15.3.6.6, (n) 15.5.7.7, (o) 15.4.3.3.1, (p) 15.4.3.5, (q) 15.4.3.4, (r) 15.5.5.4. 41. R.A. Freitas Jr., “Exploratory design in medical nanotechnology: A mechanical artificial red cell,” Artificial Cells, Blood Substitutes, and Immobil. Biotech. 26(1998):411-430; http://www.foresight.org/Nanomedicine/Respirocytes.html 42. R.A. Freitas Jr., “Microbivores: Artificial mechanical phagocytes using digest and discharge protocol,” J. Evol. Technol. 14(April 2005):1-52; http://www.jetpress.org/volume14/freitas.html; (a) 3.1.2, (b) 3.2.3, (c) 3.2.2, (d) 3.1.1, (e) 3.2.1. 43. R.A. Freitas Jr., “The future of nanofabrication and molecular scale devices in nanomedicine,” Studies in Health Technol. Inform. 80(2002):45-59. 44. R.A. Freitas Jr., “Pharmacytes: An ideal vehicle for targeted drug delivery,” J. Nanosci. Nanotechnol. 6(September/October 2006):2769-2775. 45. R.A. Freitas Jr., “Nanodentistry,” J. Amer. Dent. Assoc. 131(November 2000):1559-1566 46. R.A. Freitas Jr., Christopher J. Phoenix, “Vasculoid: A personal nanomedical appliance to replace human blood,” J. Evol. Technol. 11(April 2002):1-139; http://www.jetpress.org/volume11/vasculoid.html 47. S.L. Page, R.S. Hawley, “Chromosome choreography: the meiotic ballet,” Science 301(8 August 2003):785-789. 48. H. Hall, P. Hunt, T. Hassold, “Meiosis and sex chromosome aneuploidy: how meiotic errors cause aneuploidy; how aneuploidy causes meiotic errors,” Curr. Opin. Genet. Dev. 16(June 2006):323-329. 49. R.C. Wimalasundera, H.M. Gardiner, “Congenital heart disease and aneuploidy,” Prenat. Diagn. 24(30 December 2004):1116-1122. 50. B.A. Weaver, D.W. Cleveland, “Does aneuploidy cause cancer?” Curr. Opin. Cell Biol. 18(December 2006):658-667. 51. K.E. Drexler, Nanosystems: Molecular Machinery, Manufacturing, and Computation, John Wiley & Sons, NY, 1992; (a) 10.4.6, (b) 14.4, (c) 6.7.2, (d) 13.4, (e) Appendix A. 52. Nanofactory Collaboration website, accessed April 2007; http://www.MolecularAssembler.com/Nanofactory 53. Robert A. Freitas Jr., “Chapter 13. Progress in Nanomedicine and Medical Nanorobotics,” in Michael Rieth, Wolfram Schommers, eds., Handbook of Theoretical and Computational Nanotechnology, Volume 6 (Bioinformatics, Nanomedicine, and Drug Design), American Scientific Publishers, Stevenson Ranch, CA, 2006, pp. 619-672. 54. E.N. Marieb, Human Anatomy and Physiology, Benjamin/Cummings Publishing Company, Redwood City CA, 1989. 55. M.H. Ross, E.J. Reith, L.J. Romrell, Histology: A Text and Atlas, Second Edition, Williams & Wilkins, Baltimore MD, 1989. 56. C. Dingwall, R. Laskey, “The nuclear membrane,” Science 258(6 November 1992):942-947. 57. W.M. Becker, D.W. Deamer, The World of the Cell, Second Edition, Benjamin/Cummings Publishing Company, Redwood City CA, 1991. 58. A.J. Maniotis, C.S. Chen, D.E. Ingber, “Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure,” Proc. Natl. Acad. Sci. USA 94(February 1997):849-854. 59. L.I. Davis, “The nuclear pore complex,” Ann. Rev. Biochem. 64(1995):865-896. 60. N. Pante, U. Aebi, “Molecular dissection of the nuclear pore complex,” Crit. Rev. Biochem. Mol. Biol. 31(April 1996):153-199. 61. L.F. Pemberton, G. Blobel, J.S. Rosenblum, “Transport routes through the nuclear pore complex,” Curr. Opin. Cell Biol. 10(June 1998):392-399. 62. D. Stoffler, B. Fahrenkrog, U. Aebi, “The nuclear pore complex: From molecular architecture to functional dynamics,” Curr. Opin. Cell Biol. 11(June 1999):391-401. 63. H. Ris, “High-resolution field-emission scanning electron microscopy of nuclear pore complex,” Scanning 19(August 1997):368-375. 64. S.A. Rutherford, M.W. Goldberg, T.D. Allen, “Three-dimensional visualization of the route of protein import: the role of nuclear pore complex substructures,” Exp. Cell Res. 10(April 1997):146-160. 65. N. Pokrywka, D. Goldfarb, M. Zillmann, A. DeSilva, “The transport of macromolecules across the nuclear envelope,” in E. Edward Bittar, Neville Bittar, eds., Cellular Organelles, JAI Press, Greenwich CT, 1995, pp. 19-54. 66. E. Pennisi, “The nucleus’s revolving door,” Science 279(20 February 1998):1129-1131. 67. N. Pante, U. Aebi, “Sequential binding of import ligands to distinct nucleopore regions during their nuclear import,” Science 273(20 September 1996):1729-1732. 68. V.C. Cordes, H.R. Rackwitz, S. Reidenbach, “Mediators of nuclear protein import target karyophilic proteins to pore complexes of cytoplasmic annulate lamellae,” Exp. Cell Res. 237(15 December 1997):419-433. 69. C.L. Woodcock, “Chromatin architecture,” Curr. Opin. Struct. Biol. 16(April 2006):213-220. 70. B. Alberts, D. Bray, J. Lewis, M. Raff, K. Roberts, J.D. Watson, The Molecular Biology of the Cell, Second Edition, Garland Publishing, Inc., New York, 1989. 71. K.M. Tsutsui, K. Sano, K. Tsutsui, “Dynamic view of the nuclear matrix,” Acta Med. Okayama 59(August 2005):113-120. 72. S. Mika, B. Rost, “NMPdb: Database of Nuclear Matrix Proteins,” Nucleic Acids Res. 33(1 January 2005):D160-D163. 73. J.P. Bidwell, M. Alvarez, H. Feister, J. Onyia, J. Hock, “Nuclear matrix proteins and osteoblast gene expression,” J. Bone Miner. Res. 13(February 1998):155-167. 74. L. Hens, M. Bauman, T. Cremer, A. Sutter, J.J. Cornelis, C. Cremer, “Immunocytochemical localization of chromatin regions UV-microirradiated in S-phase or anaphase,” Exp. Cell Res. 149(1983):257-269. 75. T. Cremer, A. Kurz, R. Zirbel, S. Dietzel, B. Rinke, E. Schrock, M.R. Speicher, U. Mathieu, A. Jauch, P. Emmerich, H. Scherthan, T. Ried, C. Cremer, P. Lichter, “Role of chromosome territories in the functional compartmentalization of the cell nucleus,” Cold Spring Harbor Symp. Quant. Biol. 58(1993):777-792. 76. T. Cremer, M. Cremer, S. Dietzel, S. Muller, I. Solovei, S. Fakan, “Chromosome territories – a functional nuclear landscape,” Curr. Opin. Cell Biol. 18(June 2006):307-316. 77. N. Gilbert, S. Gilchrist, W.A. Bickmore, “Chromatin organization in the mammalian nucleus,” Int. Rev. Cytol. 242(2005):283-336. 78. L.G. Koss, “Characteristics of chromosomes in polarized normal human bronchial cells provide a blueprint for nuclear organization,” Cytogenet. Cell Genet. 82(1998):230-237. 79. D. Zink, H. Bornfleth, A. Visser, C. Cremer, T. Cremer, “Organization of early and late replicating DNA in human chromosome territories,” Exp. Cell Res. 247(25 February 1999):176-188. 80. R. Nagele, T. Freeman, L. McMorrow, H. Lee, “Precise spatial positioning of chromosomes during prometaphase: evidence for chromosomal order,” Science 270(15 December 1995):1831-1835. 81. D. Zink, T. Cremer, R. Saffrich, R. Fischer, M.F. Trendelenburg, W. Ansorge, E.H. Stelzer, “Structure and dynamics of human interphase chromosome territories in vivo,” Hum. Genet. 102(February 1998):241-251. 82. A.I. Lamond, W.C. Earnshaw, “Structure and function in the nucleus,” Science 280(24 April 1998):547-553. 83. D. Cmarko, P.J. Verschure, T.E. Martin, M.E. Dahmus, S. Krause, X.D. Fu, R. van Driel, S. Fakan, “Ultrastructural analysis of transcription and splicing in the cell nucleus after bromo-UTP microinjection,” Mol. Biol. Cell 10(1999):211-223. 84. P.J. Verschure, I. van der Kraan, E.M. Manders, R. van Driel, “Spatial relationship between transcription sites and chromosome territories,” J. Cell Biol. 147(1999):13-24. 85. A.G. Loewy, P. Siekevitz, J.R. Menninger, J.A.N. Gallant, Cell Structure and Function: An Integrated Approach, Third Edition, Saunders College Publishing, Philadelphia PA, 1991. 86. M. Mandelkern, J.G. Elias, D. Eden, D.M. Crothers, “The dimensions of DNA in solution,” J. Mol. Biol. 152(15 October 1981):153-161. 87. B. Lewin, Genes V, Oxford University Press, New York NY, 1995. 88. S.L. Wolfe, Molecular and Cellular Biology, Wadsworth Publishing Company, Belmont, California, 1993. 89. J.M. Claverie, “Gene number. What if there are only 30,000 human genes?” Science 291(16 February 2001):1255-1257. 90. National Center for Biotechnology Information, “Human Reference Sequence (from Build 33 of the Human Genome released April 14, 2003),” September 2003; http://www.ncbi.nlm.nih.gov/genome/seq/ and http://www.ornl.gov/sci/techresources/Human_Genome/posters/chromosome/faqs.shtml 91. T. Misteli, A. Gunjan, R. Hock, M. Bustin, D.T. Brown, “Dynamic binding of histone H1 to chromatin in living cells,” Nature 408(2000):877-881. 92. T. Misteli, “Protein dynamics: Implications for nuclear architecture and gene expression,” Science 291(2 February 2001):843-847. 93. R.D. Phair, T. Misteli, “High mobility of proteins in the mammalian cell nucleus,” Nature 404(2000):604-609. 94. V. Orlando, H. Strutt, R. Paro, “Analysis of chromatin structure by in vivo formaldehyde cross-linking,” Methods 11(February 1997):205-214. 95. M.D. Bennett, J.S. Heslop-Harrison, J.B. Smith, J.P. Ward, “DNA density in mitotic and meiotic metaphase chromosomes of plants and animals,” J. Cell Sci. 63(1983):173-179. 96. W. Fritzsche, E. Henderson, “Volume determination of human metaphase chromosomes by scanning force microscopy,” Scanning Microsc. 10(1996):103-110. 97. W. Fritzsche, “Salt-dependent chromosome viscoelasticity characterized by scanning force microscopy-based volume measurements,” Microsc. Res. Tech. 44(1998):357-362. 98. E.I. Kostyleva, G.V. Selivanova, I.A. Zalenskaia, “Features of the chromatin structure of erythrocytes depending on the properties of lysine-rich histones,” Mol. Biol. (Mosk) 23(January-February 1989):73-79. In Russian. 99. M. Sun, P. Serwer, “The conformation of DNA packaged in bacteriophage G,” Biophys J. 72(February 1997):958-963. 100. F.M. Richards, “Areas, volumes, packing, and protein structure,” Annu. Rev. Biophys. Bioeng. 6(1977):151-176. 101. P.C. Gasson, Geometry of Spatial Forms, John Wiley & Sons, New York, 1983. 102. J.L. Cleland, X. Lam, B. Kendrick, J. Yang, T.H. Yang, D. Overcashier, D. Brooks, C. Hsu, J.F. Carpenter, “A specific molar ratio of stabilizer to protein is required for storage stability of a lyophilized monoclonal antibody,” J. Pharm. Sci. 90(March 2001):310-321. 103. K.E. Handwerger, J.G. Gall, “Subnuclear organelles: new insights into form and function,” Trends Cell Biol. 16(January 2006):19-26. 104. D. Hernandez-Verdun, H.R. Junera, “Chapter 4. The Nucleolus,” in E.E. Bittar, N. Bittar, eds., Cellular Organelles, JAI Press, Greenwich CT, 1995, pp. 73-92. 105. A. Forer, “Chapter 23. Chromosome movements during cell-division,” in A. Lima-de-Faria, ed., Handbook of Molecular Cytology, Volume 15, North Holland Publ. Co., Amsterdam, 1969, pp. 553-601. 106. R. Rohs, I. Bloch, H. Sklenar, Z. Shakked. “Molecular flexibility in ab initio drug docking to DNA: binding-site and binding-mode transitions in all-atom Monte Carlo simulations,” Nucleic Acids Res. 33(13 December 2005):7048-7057. 107. A. Pingoud, A. Jeltsch, “Structure and function of type II restriction endonucleases,” Nucleic Acids Res. 29(15 September 2001):3705-3727. 108. P.J. Kraulis, A.R. Raine, P.L. Gadhavi, E.D. Laue, “Structure of the DNA-binding domain of zinc GAL4,” Nature 356(2 April 1992):448-450. 109. P.H. von Hippel, O.G. Berg, “Facilitated target location in biological systems,” J. Biol. Chem. 264(1989):675-678. 110. C.R. Robinson, S.G. Sligar, “Changes in solvation during DNA binding and cleavage are critical to altered specificity of the EcoRI endonuclease,” Proc. Natl Acad. Sci. (USA) 95(1998):2186-2191. 111. N.Y. Sidorova, D.C. Rau, “Differences in water release for the binding of EcoRI to specific and nonspecific DNA sequences,” Proc. Natl Acad. Sci. (USA) 93(1996):12272-12277. 112. A. Jeltsch, J. Alves, H. Wolfes, G. Maass, A. Pingoud, “Pausing of the restriction endonuclease EcoRI during linear diffusion on DNA,” Biochemistry 33(1999):10215-10219. 113. K. Grundt, L. Skjeldal, H.W. Anthonsen, T. Skauge, H.S. Huitfeldt, A.C. Ostvold, “A putative DNA-binding domain in the NUCKS protein,” Arch. Biochem. Biophys. 407(15 November 2002):168-175. 114. S. Shioi, T. Ose, K. Maenaka, M. Shiroishi, Y. Abe, D. Kohda, T. Katayama, T. Ueda, “Crystal structure of a biologically functional form of PriB from Escherichia coli reveals a potential single-stranded DNA-binding site,” Biochem. Biophys. Res. Commun. 326(28 January 2005):766-776. 115. S.X. Dou, P.Y. Wang, H.Q. Xu, X.G. Xi, “The DNA binding properties of the Escherichia coli RecQ helicase,” J. Biol. Chem. 279(20 February 2004):6354-6363. 116. D.J. Segal, C.F. Barbas 3rd, “Custom DNA-binding proteins come of age: polydactyl zinc-finger proteins,” Curr. Opin. Biotechnol. 12(December 2001):632-637. 117. J.F. Flanagan, L.Z. Mi, M. Chruszcz, M. Cymborowski, K.L. Clines, Y. Kim, W. Minor, F. Rastinejad, S. Khorasanizadeh, “Double chromodomains cooperate to recognize the methylated histone H3 tail,” Nature 438(22 December 2005):1181-1185. 118. T.M. Gottlieb, S.P. Jackson, “The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen,” Cell 72(15 January 1993):131-142. 119. S. Martensson, O. Hammarsten, “DNA-dependent protein kinase catalytic subunit. Structural requirements for kinase activation by DNA ends,” J. Biol. Chem. 277(25 January 2002):3020-3029. 120. I. Kuraoka, E.H. Morita, M. Saijo, T. Matsuda, K. Morikawa, M. Shirakawa, K. Tanaka, “Identification of a damaged-DNA binding domain of the XPA protein,” Mutat. Res. 362(2 January 1996):87-95. 121. G.W. Buchko, C.S. Tung, K. McAteer, N.G. Isern, L.D. Spicer, M.A. Kennedy, “DNA-XPA interactions: a (31)P NMR and molecular modeling study of dCCAATAACC association with the minimal DNA-binding domain (M98-F219) of the nucleotide excision repair protein XPA,” Nucleic Acids Res. 29(15 June 2001):2635-2643. 122. A.G. Cassano, V.E. Anderson, M.E. Harris, “Understanding the transition states of phosphodiester bond cleavage: insights from heavy atom isotope effects,” Biopolymers 73(January 2004):110-129. 123. N. Avvakumov, M. Sahbegovic, Z. Zhang, M. Shuen, J.S. Mymryk, “Analysis of DNA binding by the adenovirus type 5 E1A oncoprotein,” J. Gen. Virol. 83(March 2002):517-524. 124. A. Heine, J.G. Luz, C.H. Wong, I.A. Wilson, “Analysis of the class I aldolase binding site architecture based on the crystal structure of 2-deoxyribose-5-phosphate aldolase at 0.99Å resolution,” J. Mol. Biol. 343(29 October 2004):1019-1034. 125. J. Bravo, Z. Li, N.A. Speck, A.J. Warren, “The leukemia-associated AML1 (Runx1)-CBF beta complex functions as a DNA-induced molecular clamp,” Nat. Struct. Biol. 8(April 2001):371-378. 126. J.J. Hayes, R. Kaplan, K. Ura, D. Pruss, A. Wolffe, “A putative DNA binding surface in the globular domain of a linker histone is not essential for specific binding to the nucleosome,” J. Biol. Chem. 271(18 October 1996):25817-25822. 127. M. Okazaki, Y. Yoshida, S. Yamaguchi, M. Kaneno, J.C. Elliott, “Affinity binding phenomena of DNA onto apatite crystals,” Biomaterials 22(September 2001):2459-2464. 128. M. Tateno, K. Yamasaki, N. Amano, J. Kakinuma, H. Koike, M.D. Allen, M. Suzuki, “DNA recognition by beta-sheets,” Biopolymers 44(1997):335-359. 129. P. Borowski, R. Kuhl, R. Laufs, J. Schulze zur Wiesch, M. Heiland, “Identification and characterization of a histone binding site of the non-structural protein 3 of hepatitis C virus,” J. Clin. Virol. 13(June 1999):61-69. 130. T. Martin, A.M. Knapp, S. Muller, J.L. Pasquali, “Polyclonal human rheumatoid factors cross-reacting with histone H3: characterization of an idiotope on the H3 binding site,” J. Clin. Immunol. 10(July 1990):211-219. 131. O.M. Alekseev, D.C. Bencic, R.T. Richardson, E.E. Widgren, M.G. O’Rand, “Overexpression of the Linker histone-binding protein tNASP affects progression through the cell cycle,” J. Biol. Chem. 278(7 March 2003):8846-8852. 132. M.W. Trumbore, R.H. Wang, S.A. Enkemann, S.L. Berger, “Prothymosin alpha in vivo contains phosphorylated glutamic acid residues,” J. Biol. Chem. 272(17 October 1997):26394-26404. 133. M. Dobbelstein, S. Strano, J. Roth, G. Blandino, “p73-induced apoptosis: a question of compartments and cooperation,” Biochem. Biophys. Res. Commun. 331(10 June 2005):688-693. 134. “Chapter 24. Mechanics of Biological Nanotechnology,” in Bharat Bhushan, ed., Springer Handbook of Nanotechnology, Springer-Verlag, Berlin, 2004, pp. 755-760. 135. K.B. Jinesh, J.W.M. Frenken, “Capillary condensation in atomic scale friction: how water acts like a glue,” Phys. Rev. Lett. 96(2006):166103. 136. M. Egger, S.P. Heyn, H.E. Gaub, “Synthetic lipid-anchored receptors based on the binding site of a monoclonal antibody,” Biochim. Biophys. Acta 1104(17 February 1992):45-54. 137. P. Marius, S.J. Alvis, J.M. East, A.G. Lee, “The interfacial lipid binding site on the potassium channel KcsA is specific for anionic phospholipids,” Biophys. J. 89(December 2005):4081-4089. 138. C. Hornig, D. Albert, L. Fischer, M. Hornig, O. Radmark, D. Steinhilber, O. Werz, “1-Oleoyl-2-acetylglycerol stimulates 5-lipoxygenase activity via a putative (phospho)lipid binding site within the N-terminal C2-like domain,” J. Biol. Chem. 280(22 July 2005):26913-26921. 139. D. van Loon, T.A. Berkhout, R.A. Demel, K.W. Wirtz, “The lipid binding site of the phosphatidylcholine transfer protein from bovine liver,” Chem. Phys. Lipids 38(30 August 1985):29-39. 140. M.H. Lerche, B.B. Kragelund, L.M. Bech, F.M. Poulsen, “Barley lipid-transfer protein complexed with palmitoyl CoA: the structure reveals a hydrophobic binding site that can expand to fit both large and small lipid-like ligands,” Structure 5(15 February 1997):291-306. 141. T.A. Springer, J.H. Wang, “The three-dimensional structure of integrins and their ligands, and conformational regulation of cell adhesion,” Adv. Protein Chem. 68(2004):29-63. 142. G. Faury, “Role of the elastin-laminin receptor in the cardiovascular system,” Pathol. Biol. (Paris) 46(September 1998):517-526. 143. S.J. de Souza, R. Brentani, “Collagen binding site in collagenase can be determined using the concept of sense-antisense peptide interactions,” J. Biol. Chem. 267(5 July 1992):13763-13767. 144. B. Steffensen, U.M. Wallon, C.M. Overall, “Extracellular matrix binding properties of recombinant fibronectin type II-like modules of human 72-kDa gelatinase/type IV collagenase. High affinity binding to native type I collagen but not native type IV collagen,” J. Biol. Chem. 270(12 May 1995):11555-11566. 145. M. Sumper, A. Hallmann, “Biochemistry of the extracellular matrix of Volvox,” Int. Rev. Cytol. 180(1998):51-85. 146. E. Zuckerkandl, “Why so many noncoding nucleotides? The eukaryote genome as an epigenetic machine,” Genetica 115(May 2002):105-129. 147. R. Sachidanandam, D. Weissman, S.C. Schmidt, J.M. Kakol, L.D. Stein, G. Marth, S. Sherry, J.C. Mullikin, B.J. Mortimore, D.L. Willey, S.E. Hunt, C.G. Cole, P.C. Coggill, C.M. Rice, Z. Ning, J. Rogers, D.R. Bentley, P.Y. Kwok, E.R. Mardis, R.T. Yeh, B. Schultz, L. Cook, R. Davenport, M. Dante, L. Fulton, L. Hillier, R.H. Waterston, J.D. McPherson, B. Gilman, S. Schaffner, W.J. Van Etten, D. Reich, J. Higgins, M.J. Daly, B. Blumenstiel, J. Baldwin, N. Stange-Thomann, M.C. Zody, L. Linton, E.S. Lander, D. Altshuler, “International SNP Map Working Group. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms,” Nature 409(15 February 2001):928-933. 148. W.P. Wahls, P.D. Moore, “Recombination hotspot activity of hypervariable minisatellite DNA requires minisatellite DNA binding proteins,” Somat. Cell Mol. Genet. 24(January 1998):41-51. 149. S. Myers, L. Bottolo, C. Freeman, G. McVean, P. Donnelly, “A fine-scale map of recombination rates and hotspots across the human genome,” Science 310(14 October 2005):321-324. 150. G.A. Calin, C.M. Croce, “MicroRNAs and chromosomal abnormalities in cancer cells,” Oncogene 25(9 October 2006):6202-6210. 151. L. Pasqualucci, P. Neumeister, T. Goossens, G. Nanjangud, R.S. Chaganti, R. Kuppers, R. Dalla-Favera, “Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas,” Nature 412(19 July 2001):341-346. 152. C. Yauk, “Monitoring for induced heritable mutations in natural populations: application of minisatellite DNA screening,” Mutat. Res. 411(August 1998):1-10. 153. H.E. Young, “Existence of reserve quiescent stem cells in adults, from amphibians to humans,” Curr. Top. Microbiol. Immunol. 280(2004):71-109. 154. R. Pelayo, K. Miyazaki, J. Huang, K.P. Garrett, D.G. Osmond, P.W. Kincade, “Cell cycle quiescence of early lymphoid progenitors in adult bone marrow,” Stem Cells 24(December 2006):2703-2713. 155. E. Santoni-Rugiu, P. Jelnes, S.S. Thorgeirsson, H.C. Bisgaard, “Progenitor cells in liver regeneration: molecular responses controlling their activation and expansion,” APMIS 113(November-December 2005):876-902. 156. F. Relaix, “Skeletal muscle progenitor cells: from embryo to adult,” Cell Mol. Life Sci. 63(June 2006):1221-1225. 157. H.J. Klassen, K.L. Imfeld, I.I. Kirov, L. Tai, F.H. Gage, M.J. Young, M.A. Berman, “Expression of cytokines by multipotent neural progenitor cells,” Cytokine 22(May 2003):101-106. 158. A. Armakolas, A.J. Klar, “Cell type regulates selective segregation of mouse chromosome 7 DNA strands in mitosis,” Science 311(24 February 2006):1146-1149. 159. E. Tuzun, A.J. Sharp, J.A. Bailey, R. Kaul, V.A. Morrison, L.M. Pertz, E. Haugen, H. Hayden, D. Albertson, D. Pinkel, M.V. Olson, E.E. Eichler, “Fine-scale structural variation of the human genome,” Nat. Genet. 37(July 2005):727-732.
160. J. Sebat, B. Lakshmi, J. Troge, J. Alexander, J. Young, P. Lundin, S. Maner, H. Massa, M. Walker, M. Chi, N. Navin, R. Lucito, J. Healy, J. Hicks, K. Ye, A. Reiner, T.C. Gilliam, B. Trask, N. Patterson, A. Zetterberg, M. Wigler, “Large-scale copy number polymorphism in the human genome,” Science 305(23 July 2004):525-528. 161. L. Feuk, A.R. Carson, S.W. Scherer, “Structural variation in the human genome,” Nat. Rev. Genet. 7(February 2006):85-97. 162. R. Redon, S. Ishikawa, K.R. Fitch, L. Feuk, G.H. Perry, T.D. Andrews, H. Fiegler, M.H. Shapero, A.R. Carson, W. Chen, E.K. Cho, S. Dallaire, J.L. Freeman, J.R. Gonzalez, M. Gratacos, J. Huang, D. Kalaitzopoulos, D. Komura, J.R. MacDonald, C.R. Marshall, R. Mei, L. Montgomery, K. Nishimura, K. Okamura, F. Shen, M.J. Somerville, J. Tchinda, A. Valsesia, C. Woodwark, F. Yang, J. Zhang, T. Zerjal, J. Zhang, L. Armengol, D.F. Conrad, X. Estivill, C. Tyler-Smith, N.P. Carter, H. Aburatani, C. Lee, K.W. Jones, S.W. Scherer, M.E. Hurles, “Global variation in copy number in the human genome,” Nature 444(23 November 2006):444-454. 163. D. Komura, F. Shen, S. Ishikawa, K.R. Fitch, W. Chen, J. Zhang, G. Liu, S. Ihara, H. Nakamura, M.E. Hurles, C. Lee, S.W. Scherer, K.W. Jones, M.H. Shapero, J. Huang, H. Aburatani, “Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays,” Genome Res. 16(December 2006):1575-1584. 164. R.E. Mills, C.T. Luttig, C.E. Larkins, A. Beauchamp, C. Tsui, W.S. Pittard, S.E. Devine, “An initial map of insertion and deletion (INDEL) variation in the human genome,” Genome Res. 16(September 2006):1182-1190. 165. T. Haaf, “Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development,” Curr. Top. Microbiol. Immunol. 310(2006):13-22. 166. I.C. Weaver, N. Cervoni, F.A. Champagne, A.C. D’Alessio, S. Sharma, J.R. Seckl, S. Dymov, M. Szyf, M.J. Meaney, “Epigenetic programming by maternal behavior,” Nat. Neurosci. 7(August 2004):847-854. 167. R.Z. Chen, U. Pettersson, C. Beard, L. Jackson-Grusby, R. Jaenisch, “DNA hypomethylation leads to elevated mutation rates,” Nature 395(3 September 1998):89-93. 168. M. Nakayama, M.L. Gonzalgo, S. Yegnasubramanian, X. Lin, A.M. De Marzo, W.G. Nelson, “GSTP1 CpG island hypermethylation as a molecular biomarker for prostate cancer,” J. Cell Biochem. 91(15 February 2004):540-552. 169. J.C. Chow, Z. Yen, S.M. Ziesche, C.J. Brown, “Silencing of the mammalian X chromosome,” Annu. Rev. Genomics Hum. Genet. 6(2005):69-92. 170. U.R. Anoop, V. Ramesh, P.D. Balamurali, O. Nirima, B. Premalatha, V.P. Karthikshree, “Role of Barr bodies obtained from oral smears in the determination of sex,” Indian J. Dent. Res. 15(January-March 2004):5-7. 171. D. Haig, “Self-imposed silence: parental antagonism and the evolution of X-chromosome inactivation,” Evolution Int. J. Org. Evolution 60(March 2006):440-447. 172. R.P. Tucker, D. Kenzelmann, A. Trzebiatowska, R. Chiquet-Ehrismann, “Teneurins: Transmembrane proteins with fundamental roles in development,” Int. J. Biochem. Cell Biol. 39(2007):292-297. 173. K.H. Orstavik, “Skewed X inactivation in healthy individuals and in different diseases,” Acta Paediatr. Suppl. 95(April 2006):24-29. 174. Z.E. Karanjawala, M.R. Lieber, “DNA damage and aging,” Mech. Ageing Dev. 125(June 2004):405-416. 175. M. Dizdaroglu, P. Jaruga, M. Birincioglu, H. Rodriguez, “Free radical-induced damage to DNA: mechanisms and measurement,” Free Radic. Biol. Med. 32(1 June 2002):1102-1115. 176. M. Islaih, B.W. Halstead, I.A. Kadura, B. Li, J.L. Reid-Hubbard, L. Flick, J.L. Altizer, J. Thom Deahl, D.K. Monteith, R.K. Newton, D.E. Watson, “Relationships between genomic, cell cycle, and mutagenic responses of TK6 cells exposed to DNA damaging chemicals,” Mutat. Res. 578(15 October 2005):100-116. 177. W. Heywood, B. Henderson, S.P. Nair, “Cytolethal distending toxin: creating a gap in the cell cycle,” J. Med. Microbiol. 54(March 2005):207-216. 178. A. Sinclair, S. Yarranton, C. Schelcher, “DNA-damage response pathways triggered by viral replication,” Expert Rev. Mol. Med. 8(3 March 2006):1-11. 179. W.C. Hahn, M. Meyerson, “Telomerase activation, cellular immortalization and cancer,” Ann. Med. 33(March 2001):123-129. 180. J. Yang, E. Chang, A.M. Cherry, C.D. Bangs, Y. Oei, A. Bodnar, A. Bronstein, C.P. Chiu, G.S. Herron, “Human endothelial cell life extension by telomerase expression,” J. Biol. Chem. 274(10 September 1999):26141-26148. 181. A. Ahmed, T.O. Tollefsbol, “Telomerase, telomerase inhibition, and cancer,” J. Anti Aging Med. 6(2003):315-325. 182. J. Mitteldorf, “How evolutionary thinking affects people's ideas about aging interventions,” Rejuvenation Res. 9(Summer 2006):346-350. 183. W.T. O’Donnell, S.T. Warren, “A decade of molecular studies of fragile X syndrome,” Annu. Rev. Neurosci. 25(2002):315-338. 184. N. de Parseval, T. Heidmann, “Human endogenous retroviruses: from infectious elements to human genes,” Cytogenet. Genome Res. 110(2005):318-332. 185. H.T. Bjornsson, L.M. Ellingsen, J.J. Jonsson, “Transposon-derived repeats in the human genome and 5-methylcytosine-associated mutations in adjacent genes,” Gene 370(29 March 2006):43-50. 186. H. Debrauwere, J. Buard, J. Tessier, D. Aubert, G. Vergnaud, A. Nicolas, “Meiotic instability of human minisatellite CEB1 in yeast requires DNA double-strand breaks,” Nat. Genet. 23(November 1999):367-371. 187. R.E. Mills, E.A. Bennett, R.C. Iskow, C.T. Luttig, C. Tsui, W.S. Pittard, S.E. Devine, “Recently mobilized transposons in the human and chimpanzee genomes,” Am. J. Hum. Genet. 78(April 2006):671-679. 188. J. Lee, R. Cordaux, K. Han, J. Wang, D.J. Hedges, P. Liang, M.A. Batzer, “Different evolutionary fates of recently integrated human and chimpanzee LINE-1 retrotransposons,” Gene 390(1 April 2007):18-27. 189. N. Gilbert, S. Lutz-Prigge, J.V. Moran, “Genomic deletions created upon LINE-1 retrotransposition,” Cell 110(9 August 2002):315-325. 190. D.E. Symer, C. Connelly, S.T. Szak, E.M. Caputo, G.J. Cost, G. Parmigiani, J.D. Boeke, “Human L1 retrotransposition is associated with genetic instability in vivo,” Cell 110(9 August 2002):327-38. 191. W.A. Schulz, “L1 retrotransposons in human cancers,” J. Biomed. Biotechnol. 2006:83672.
192. R. Cordaux, J. Lee, L. Dinoso, M.A. Batzer, “Recently integrated Alu retrotransposons are essentially neutral residents of the human genome,” Gene 373(24 May 2006):138-144.
193. C. Wyman, D. Ristic, R. Kanaar, “Homologous recombination-mediated double-strand break repair,” DNA Repair (Amst) 3(August-September 2004):827-833.
194. M. van den Bosch, P.H. Lohman, A. Pastink, “DNA double-strand break repair by homologous recombination,” Biol. Chem. 383(June 2002):873-892.
195. R.W. Van Nues, J.D. Brown, “Saccharomyces SRP RNA secondary structures: a conserved S-domain and extended Alu-domain,” RNA 10(January 2004):75-89.
196. O. Weichenrieder, U. Kapp, S. Cusack, K. Strub, “Identification of a minimal Alu RNA folding domain that specifically binds SRP9/14,” RNA 3(November 1997):1262-1274.
197. P. Hilgard, T. Huang, A.W. Wolkoff, R.J. Stockert, “Translated Alu sequence determines nuclear localization of a novel catalytic subunit of casein kinase 2,” Am. J. Physiol. Cell Physiol. 283(August 2002):C472-C483.
198. K. Sobczak, W.J. Krzyzosiak, “Structural determinants of BRCA1 translational regulation,” J. Biol. Chem. 277(10 May 2002):17349-17358. 199. J.H. Hanke, J.E. Hambor, P. Kavathas, “Repetitive Alu elements form a cruciform structure that regulates the function of the human CD8 alpha T cell-specific enhancer,” J. Mol. Biol. 246(10 February 1995):63-73. 200. C. Ofria, C. Adami, T.C. Collier, “Selective pressures on genomes in molecular evolution,” J. Theor. Biol. 222(21 June 2003):477-483. 201. S.M. Gasser, “Visualizing chromatin dynamics in interphase nuclei,” Science 296(24 May 2000):1412-1416. 202. J. Coulombe-Huntington, J. Majewski, “Characterization of intron loss events in mammals,” Genome Res. 17(January 2007):23-32. 203. K. Hu, “Intron exclusion and the mystery of intron loss,” FEBS Lett. 580(27 November 2006):6361-6365. 204. L.C. Wong, L.F. Landweber, “Evolution of programmed DNA rearrangements in a scrambled gene,” Mol. Biol. Evol. 23(April 2006):756-763. 205. G.W. Rebeck, M.J. Ladu, S. Estus, G. Bu, E.J. Weeber, “The generation and function of soluble apoE receptors in the CNS,” Mol. Neurodegener. 1(24 October 2006):15. 206. Y. Huang, K.H. Weisgraber, L. Mucke, R.W. Mahley, “Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer’s disease,” J. Mol. Neurosci. 23(2004):189-204. 207. R.W. Mahley, K.H. Weisgraber, Y. Huang, “Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease,” Proc. Natl. Acad. Sci. (USA) 103(11 April 2006):5644-5651. 208. “Genome Pioneer Will Start Center of His Own,” The New York Times, 15 August 2002; http://www.kurzweilai.net/news/frame.html?main=/news/news_single.html?id%3D1180 209. K. Davies, “Venter Raises Stakes for $1,000 Genome Prize,” Bio-IT World, 19 October 2005; http://www.bio-itworld.com/newsitems/2005/oct2005/10-19-05-news-genome-prize 210. M. Anderson, “NIH offers $1000 genome grant,” The Scientist 5(2004):20040223-05; http://www.the-scientist.com/news/20040223/05/ 211. A. Meller, L. Nivon, E. Brandin, J. Golovchenko, D. Branton, “Rapid nanopore discrimination between single polynucleotide molecules,” Proc. Natl. Acad. Sci. (USA) 97(1 February 2000):1079-1084; http://www.pnas.org/cgi/content/full/97/3/1079 212. A. Meller, L. Nivon, D. Branton, “Voltage-driven DNA translocations through a nanopore,” Phys. Rev. Lett. 86(2001):3435-3438. 213. A. Meller, D. Branton, “Single molecule measurements of DNA transport through a nanopore,” Electrophoresis 23(2002):2583-2591; http://mcb.harvard.edu/branton/MellerBranton04.pdf 214. D.W. Deamer, D. Branton, “Characterization of nucleic acids by nanopore analysis,” Acc. Chem. Res. 35(2002):817-825. 215. D. Branton, A. Meller, “Using nanopores to discriminate between single molecules of DNA,” in J.J. Kasianowicz, M.S.Z. Kellermayer, D.W. Deamer, eds., Structure and Dynamics of Confined Polymers, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2002, pp. 177-185; http://mcb.harvard.edu/branton/BrantonMeller02.pdf 216. H. Wang, D. Branton, “Nanopores with a spark for single-molecule detection,” Nature Biotechnol. 19(2001):622-623; http://mcb.harvard.edu/branton/WangBrantonNewsViews01.Actual.pdf 217. J. Li, M. Gershow, D. Stein, E. Brandin, J.A. Golovchenko, “DNA molecules and configurations in a solid-state nanopore microscope,” Nature Mater. 2(2003):611-615; http://mcb.harvard.edu/branton/LiGershowSteinOnlinePub.pdf 218. H. Wang, J.E. Dunning, A.P. Huang, J.A. Nyamwanda, D. Branton, “DNA heterogeneity and phosphorylation unveiled by single-molecule electrophoresis,” Proc. Natl. Acad. Sci. (USA) 101(14 September 2004):13472-13477. 219. J. Li, D. Stein, C. McMullan, D. Branton, M.J. Aziz, J.A. Golovchenko, “Ion-beam sculpting at nanometre length scales,” Nature 412(2001):166-169; http://mcb.harvard.edu/branton/LiNature01.Actual.pdf 220. D. Stein, J. Li, J.A. Golovchenko, “Ion-beam sculpting time scales,” Phys. Rev. Lett. 89(2002):276106; http://mcb.harvard.edu/branton/SteinLiGolovchenko.pdf 221. A.J. Storm, J.H. Chen, X.S. Ling, H.W. Zandbergen, C. Dekker, “Fabrication of solid-state nanopores with single-nanometre precision,” Nat. Mater. 2(August 2003):537-540. 222. A.F. Sauer-Budge, J.A. Nyamwanda, D.K. Lubensky, D. Branton, “Unzipping kinetics of double-stranded DNA in a nanopore,” Phys. Rev. Lett. 90(2003):238101; http://mcb.harvard.edu/branton/Sauer-BudgePhysicalReviewLetters.pdf 223. D.W. Deamer, M. Akeson, “Nanopores and nucleic acids: prospects for ultrarapid sequencing,” Trends Biotechnol. 18(April 2000):147-151. 224. J. Lagerqvist, M. Zwolak, M. Di Ventra, “Fast DNA sequencing via transverse electronic transport,” Nano Lett. 6(April 2006):779-782. 225. S. Iwabuchii, T. Mori, K. Ogawa, K. Sato, M. Saito, Y. Morita, T. Ushiki, E. Tamiya, “Atomic force microscope-based dissection of human metaphase chromosomes and high resolutional imaging by carbon nanotube tip,” Arch. Histol. Cytol. 65(December 2002):473-479. 226. M. Oberringer, A. Englisch, B. Heinz, H. Gao, T. Martin, U. Hartmann, “Atomic force microscopy and scanning near-field optical microscopy studies on the characterization of human metaphase chromosomes,” Eur. Biophys. J. 32(November 2003):620-627. 227. D. Fukushi, T. Ushiki, “The structure of C-banded human metaphase chromosomes as observed by atomic force microscopy,” Arch. Histol. Cytol. 68(2005):81-87. 228. H.J. An, Y.C. Guo, X.D. Zhang, Y. Zhang, J. Hu, “Nanodissection of single- and double-stranded DNA by atomic force microscopy,” J. Nanosci. Nanotechnol. 5(October 2005):1656-1659. 229. O. Hoshi, M. Shigeno, T. Ushiki, “Atomic force microscopy of native human metaphase chromosomes in a liquid,” Arch. Histol. Cytol. 69(March 2006):73-78. 230. S. Thalhammer, R.W. Stark, S. Muller, J. Wienberg, W.M. Heckl, “The atomic force microscope as a new microdissecting tool for the generation of genetic probes,” J. Struct. Biol. 119(July 1997):232-237. 231. K. Konig, I. Riemann, P. Fischer, K.J. Halbhuber, “Intracellular nanosurgery with near infrared femtosecond laser pulses,” Cell Mol. Biol. 45(March 1999):195-201. 232. I. Kelson, S. Nussinov, “A scheme for sequencing large DNA molecules by identifying local nuclear-induced effects,” Proc. Natl. Acad. Sci. (USA) 91(19 July 1994):6963-6966. 233. G.H. Seong, T. Niimi, Y. Yanagida, E. Kobatake, M. Aizawa, “Single-molecular AFM probing of specific DNA sequencing using RecA-promoted homologous pairing and strand exchange,” Anal. Chem. 72(15 March 2000):1288-1293. 234. N.K. Voulgarakis, A. Redondo, A.R. Bishop, K.O. Rasmussen, “Sequencing DNA by dynamic force spectroscopy: limitations and prospects,” Nano Lett. 6(July 2006):1483-1486. 235. S. Cocco, R. Monasson, J.F. Marko, “Unzipping dynamics of long DNAs,” Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 66(November 2002):051914. 236. B.D. Sattin, A.E. Pelling, M.C. Goh, “DNA base pair resolution by single molecule force spectroscopy,” Nucleic Acids Res. 32(14 September 2004):4876-4883. 237. N. Singh, Y. Singh, “Effect of genome sequence on the force-induced unzipping of a DNA molecule,” Eur. Phys. J. E Soft Matter 19(February 2006):233-238. 238. C.H. Lee, C. Danilowicz, V.W. Coljee, M. Prentiss, “Comparison of the measured phase diagrams in the force-temperature plane for the unzipping of two different natural DNA sequences,” Eur. Phys. J. E Soft Matter 19(March 2006):339-344. 239. V. Baldazzi, S. Cocco, E. Marinari, R. Monasson, “Inference of DNA sequences from mechanical unzipping: an ideal-case study,” Phys. Rev. Lett. 96(31 March 2006):128102. 240. I.M. Wilson, J.J. Davies, M. Weber, C.J. Brown, C.E. Alvarez, C. MacAulay, D. Schubeler, W.L. Lam, “Epigenomics: mapping the methylome,” Cell Cycle 5(January 2006):155-158.
241. A. Jeltsch, J. Walter, R. Reinhardt, M. Platzer, “German human methylome project started,” Cancer Res. 66(15 July 2006):7378.
242. H. Schob, U. Grossniklaus, “The first high-resolution DNA “methylome”, Cell 126(22 September 2006):1025-1028. 243. Ralph C. Merkle, “Convergent assembly,” Nanotechnology 8(1997):18-22; http://www.zyvex.com/nanotech/convergent.html 244. Robert A. Freitas Jr., Ralph C. Merkle, Kinematic Self-Replicating Machines, Landes Bioscience, Georgetown, TX, 2004; http://www.MolecularAssembler.com/KSRM.htm 245. K.H. Nierhaus, D.N. Wilson, eds., Protein Synthesis and Ribosome Structure: Translating the Genome, John Wiley & Sons NY, 2004. 246. T. Lionnet, A. Dawid, S. Bigot, F.X. Barre, O.A. Saleh, F. Heslot, J.F. Allemand, D. Bensimon, V. Croquette, “DNA mechanics as a tool to probe helicase and translocase activity,” Nucleic Acids Res. 34(2006):4232-4244. 247. A.-S. Duwez, S. Cuenot, C. Jerome, S. Gabriel, R. Jerome, S. Rapino, F. Zerbetto, “Mechanochemistry: targeted delivery of single molecules,” Nature Nanotechnology 1(2006):122-125. 248. J. Peng, R.A. Freitas Jr., R.C. Merkle, J.R. Von Ehr, J.N. Randall, G.D. Skidmore, “Theoretical Analysis of Diamond Mechanosynthesis. Part III. Positional C2 deposition on diamond C(110) surface using Si/Ge/Sn-based dimer placement tools,” J. Comput. Theor. Nanosci. 3(February 2006):28-41. 249. B. Temelso, C.D. Sherrill, R.C. Merkle, R.A. Freitas Jr., “High-level ab initio studies of hydrogen abstraction from prototype hydrocarbon systems,” J. Phys. Chem. A 110 (28 September 2006):11160-11173. 250. R.A. Freitas Jr., D.G. Allis, R.C. Merkle, “Horizontal Ge-substituted polymantane-based C2 dimer placement tooltip motifs for diamond mechanosynthesis,” J. Comput. Theor. Nanosci. 4(2007). In press. 251. Custom DNA and peptide sequence Web ordering; see: http://www.sigmaaldrich.com/Brands/Sigma_Genosys.html; http://www.biosyn.com/order.htm; and http://mbcf.dfci.harvard.edu/docs/DNAcomp.html
252. H. Koster, H. Blocker, R. Frank, S. Geussenhainer, W. Kaiser, “Total synthesis of a structural gene for the human peptide hormone angiotensin II,” Hoppe Seylers Z. Physiol. Chem. 356(October 1975):1585-1593.
253. R. Crea, A. Kraszewski, T. Hirose, K. Itakura, “Chemical synthesis of genes for human insulin,” Proc. Natl. Acad. Sci. (USA) 75(December 1978):5765-5769. 254. H.G. Khorana, “Total synthesis of a gene,” Science 203(16 February 1979):614-625. 255. M.D. Edge, A.R. Green, G.R. Heathcliffe, P.A. Meacock, W. Schuch, D.B. Scanlon, T.C. Atkinson, C.R. Newton, A.F. Markham, “Total synthesis of a human leukocyte interferon gene,” Nature 292(20 August 1981):756-762. 256. J.A. Tang, X.X. Zhou, Z.H. Liang, B.Y. Zou, X. Gao, X.Y. Yang, C.Q. Chen, D.B. Wang, “Synthesis of the leu-enkephalin gene,” Sci. Sin. B 27(May 1984):466-477. 257. L. Ferretti, S.S. Karnik, H.G. Khorana, M. Nassal, D.D. Oprian, “Total synthesis of a gene for bovine rhodopsin,” Proc. Natl. Acad. Sci. (USA) 83(February 1986):599-603. 258. M.A. Wosnick, R.W. Barnett, A.M. Vicentini, H. Erfle, R. Elliott, M. Sumner-Smith, N. Mantei, R.W. Davies, “Rapid construction of large synthetic genes: total chemical synthesis of two different versions of the bovine prochymosin gene,” Gene 60(1987):115-127. 259. Z.A. Shabarova, I.N. Merenkova, T.S. Oretskaya, N.I. Sokolova, E.A. Skripkin, E.V. Alexeyeva, A.G. Balakin, A.A. Bogdanov, “Chemical ligation of DNA: the first non-enzymatic assembly of a biologically active gene,” Nucleic Acids Res. 19(11 August 1991):4247-4251. 260. Y. Harada, T. Sakamoto, T. Shinomura, K. Takamoto, T. Senda, M. Tsuda, “Total synthesis of a gene for octopus rhodopsin and its preliminary expression,” J. Biochem. (Tokyo) 110(October 1991):501-507. 261. W.P. Stemmer, A. Crameri, K.D. Ha, T.M. Brennan, H.L. Heyneker, “Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides,” Gene 164(16 October 1995):49-53. 262. W.H. Attatippaholkun, M.K. Attatippaholkun, A. Nisalak, D.W. Vaughn, B.L. Innis, “A novel method for the preparation of large cDNA fragments from dengue-3 RNA genome by long RT-PCR amplification,” Southeast Asian J. Trop. Med. Public Health 31(2000):126-133 (Suppl 1). 263. D.M. Hoover, J. Lubkowski, “DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis,” Nucleic Acids Res. 30(15 May 2002):e43. 264. J.M. Rouillard, W. Lee, G. Truan, X. Gao, X. Zhou, E. Gulari, “Gene2Oligo: oligonucleotide design for in vitro gene synthesis,” Nucleic Acids Res. 32(1 July 2004):W176-W180. 265. A.S. Xiong, Q.H. Yao, R.H. Peng, X. Li, H.Q. Fan, Z.M. Cheng, Y. Li, “A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences,” Nucleic Acids Res. 32(7 July 2004):e98. 266. J. Tian, H. Gong, N. Sheng, X. Zhou, E. Gulari, X. Gao, G. Church, “Accurate multiplex gene synthesis from programmable DNA microchips,” Nature 432(23 December 2004):1050-1054. 267. H.G. Menzella, S.J. Reisinger, M. Welch, J.T. Kealey, J. Kennedy, R. Reid, C.Q. Tran, D.V. Santi, “Redesign, synthesis and functional expression of the 6-deoxyerythronolide B polyketide synthase gene cluster,” J. Ind. Microbiol. Biotechnol. 33(January 2006):22-28. 268. “Researchers build huge DNA chains,” BBC News, Sci/Tech Archives, 27 January 2000; http://news.bbc.co.uk/1/hi/sci/tech/620847.stm 269. H.O. Smith, C.A. Hutchison 3rd, C. Pfannkoch, J. Craig Venter, “Generating a synthetic genome by whole genome assembly: {phi}X174 bacteriophage from synthetic oligonucleotides,” Proc. Natl. Acad. Sci. (USA) 100(23 December 2003):15440-15445. 270. G.A. Evans (Egea Biosciences, Inc., San Diego, CA), “Method for the complete chemical synthesis and assembly of genes and genomes,” United States Patent No. 6,521,427, 18 February 2003. 271. X. Zhou, S. Cai, A. Hong, Q. You, P. Yu, N. Sheng, O. Srivannavit, S. Muranjan, J.M. Rouillard, Y. Xia, X. Zhang, Q. Xiang, R. Ganesh, Q. Zhu, A. Matejko, E. Gulari, X. Gao, “Microfluidic PicoArray synthesis of oligodeoxynucleotides and simultaneous assembling of multiple DNA sequences,” Nucleic Acids Res. 32(11 October 2004):5409-5417. 272. N.A. Shevchuk, A.V. Bryksin, Y.A. Nusinovich, F.C. Cabello, M. Sutherland, S. Ladisch, “Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously,” Nucleic Acids Res. 32(22 January 2004):e19. 273. S.J. Kodumal, J.G. Patel, R. Reid, H.G. Menzella, M. Welch, D.V. Santi, “Total synthesis of long DNA sequences: synthesis of a contiguous 32-kb polyketide synthase gene cluster,” Proc. Natl. Acad. Sci. (USA) 101(2 November 2004):15573-15578. 274. A.C. Forster, G.M. Church, “Towards synthesis of a minimal cell,” Mol. Syst. Biol. 2(2006):45. 275. I. Taylor, D. Watts, G. Kneale, “Substrate recognition and selectivity in the type IC DNA modification methylase M.EcoR124I,” Nucleic Acids Res. 21(25 October 1993):4929-4935. 276. V.A. Chernukhin, Y.G. Kashirina, K.S. Sukhanova, M.A. Abdurashitov, D.A Gonchar, S.Kh. Degtyarev, “Isolation and characterization of biochemical properties of DNA methyltransferase FauIA modifying the second cytosine in the nonpalindromic sequence 5’-CCCGC-3’,” Biochemistry (Mosc) 70(June 2005):685-691. 277. P.A. Carr, J.S. Park, Y.J. Lee, T. Yu, S. Zhang, J.M. Jacobson, “Protein-mediated error correction for de novo DNA synthesis,” Nucleic Acids Res. 32(23 November 2004):e162. 278. B.F. Binkowski, K.E. Richmond, J. Kaysen, M.R. Sussman, P.J. Belshaw, “Correcting errors in synthetic DNA through consensus shuffling,” Nucleic Acids Res. 33(30 March 2005):e55. 279. M. Fuhrmann, W. Oertel, P. Berthold, P. Hegemann, “Removal of mismatched bases from synthetic genes by enzymatic mismatch cleavage,” Nucleic Acids Res. 33(30 March 2005):e58. 280. I. Cserpan, M. Kalman, M.L. Tjornhammar, A. Simoncsits, “Conversion of single-stranded oligonucleotides into cloned duplexes and its consecutive application to short artificial genes,” Acta Chem. Scand. 45(March 1991):265-272. 281. D. Nykypanchuk, H.H. Strey, D.A. Hoagland, “Brownian motion of DNA confined within a two-dimensional array,” Science 297(9 August 2002):987-990. 282. S. Bheemanaik, Y.V. Reddy, D.N. Rao, “Structure, function and mechanism of exocyclic DNA methyltransferases,” Biochem J. 399(15 October 2006):177-190.
283. G. Pljevaljcic, F. Schmidt, A. Peschlow, E. Weinhold, “Sequence-specific DNA labeling using methyltransferases,” Methods Mol Biol. 283(2004):145-161.
284. C. Dalhoff, G. Lukinavicius, S. Klimasauskas, E. Weinhold, “Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases,” Nat. Chem. Biol. 2(January 2006):31-32. 285. H.A. LaVoie, “Epigenetic control of ovarian function: the emerging role of histone modifications,” Mol. Cell. Endocrinol. 243(24 November 2005):12-18. 286. A.L. Clayton, C.A. Hazzalin, L.C. Mahadevan, “Enhanced histone acetylation and transcription: a dynamic perspective,” Mol. Cell 23(4 August 2006):289-296. 287. M.S. Torok, P.A. Grant, “The generation and recognition of histone methylation,” Results Probl. Cell. Differ. 41(2006):25-46. 288. Y. Wang, W. Fischle, W. Cheung, S. Jacobs, S. Khorasanizadeh, C.D. Allis, “Beyond the double helix: writing and reading the histone code,” Novartis Found. Symp. 259(2004):3-17. 289. M.A. Osley, “H2B ubiquitylation: the end is in sight,” Biochim. Biophys. Acta 1677(15 March 2004):74-78. 290. D. Nathan, K. Ingvarsdottir, D.E. Sterner, G.R. Bylebyl, M. Dokmanovic, J.A. Dorsey, K.A. Whelan, M. Krsmanovic, W.S. Lane, P.B. Meluh, E.S. Johnson, S.L. Berger, “Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications,” Genes Dev. 20(15 April 2006):966-976. 291. A. Villar-Garea, A. Imhof, “The analysis of histone modifications,” Biochim. Biophys. Acta 1764(December 2006):1932-1939. 292. D.V. Fyodorov, J.T. Kadonaga, “Dynamics of ATP-dependent chromatin assembly by ACF,” Nature 418(22 August 2002):896-900. 293. M.-A. Hakimi, D.A. Bochar, J.A. Schmiesing, Y. Dong, O.G. Barak, D.W. Speicher, K. Yokomori, R. Shiekhattar, “A chromatin remodelling complex that loads cohesin onto human chromosomes,” Nature 418(29 August 2002):994-998. 294. D.R. Foltz, L.E. Jansen, B.E. Black, A.O. Bailey, J.R. Yates 3rd, D.W. Cleveland, “The human CENP-A centromeric nucleosome-associated complex,” Nat. Cell. Biol. 8(May 2006):458-469. 295. P.G. Prasanna, W.F. Blakely, “Premature chromosome condensation in human resting peripheral blood lymphocytes for chromosome aberration analysis using specific whole-chromosome DNA hybridization probes,” Methods Mol. Biol. 291(2005):49-57. 296. K. Kimura, V.V. Rybenkov, N.J. Crisona, T. Hirano, N.R. Cozzarelli, “13S condensin actively reconfigures DNA by introducing global positive writhe: implications for chromosome condensation,” Cell 98(23 July 1999):239-248. 297. S.A. Grigoryev, J. Bednar, C.L. Woodcock, “MENT, a heterochromatin protein that mediates higher order chromatin folding, is a new serpin family member,” J. Biol. Chem. 274(26 February 1999):5626-5636. 298. P. Guo, “Bacterial virus phi29 DNA-packaging motor and its potential applications in gene therapy and nanotechnology,” Methods Mol. Biol. 300(2005):285-324. 299. C. Forrey, M. Muthukumar, “Langevin dynamics simulations of genome packing in bacteriophage,” Biophys. J. 91(1 July 2006):25-41. 300. H.G. Garcia, P. Grayson, L. Han, M. Inamdar, J. Kondev, P.C. Nelson, R. Phillips, J. Widom, P.A. Wiggins, “Biological consequences of tightly bent DNA: The other life of a macromolecular celebrity,” Biopolymers 85(5 February 2007):115-130. 301. P. Razzell, “Smallpox extinction – a note of caution,” New Scientist 71(1976):35. 302. F.O. MacCallum, J.R. McDonald, “Survival of variola virus in raw cotton,” Bull. World Health Organ. 16(1957):247-254. 303. H.L. Wolff, J.J. Croon, “The survival of smallpox virus (variola minor) in natural circumstances,” Bull. World Health Organ. 38(1968):492-493. 304. M.R. Prausnitz, “Microneedles for transdermal drug delivery,” Adv. Drug Deliv. Rev. 56(27 March 2004):581-587. 305. J.B. Christoforidis, S.M. Warden, M. Demattia, D.J. D’Amico, “In vivo redirection of retinal blood flow into borosilicate micro-cannulas in rabbits,” Graefes Arch. Clin. Exp. Ophthalmol. 244(August 2006):1022-1025. 306. T.W. Olsen, X. Feng, K. Wabner, S.R. Conston, D.H. Sierra, D.V. Folden, M.E. Smith, J.D. Cameron, “Cannulation of the suprachoroidal space: a novel drug delivery methodology to the posterior segment,” Am. J. Ophthalmol. 142(November 2006):777-787. 307. M.G. Kolonin, J. Sun, K.A. Do, C.I. Vidal, Y. Ji, K.A. Baggerly, R. Pasqualini, W. Arap, “Synchronous selection of homing peptides for multiple tissues by in vivo phage display,” FASEB J. 20(May 2006):979-981. 308. B. Petri, M.G. Bixel, “Molecular events during leukocyte diapedesis,” FEBS J. 273(October 2006):4399-4407. 309. J.V. Hurley, “An electron microscopic study of leucocytic emigration and vascular permeability in rat skin,” Austral. J. Exp. Biol. 41(1963):171-186. 310. J.P. Olano, “Rickettsial infections,” Ann. N.Y. Acad. Sci. 1063(December 2005):187-196. 311. R.A. Heinzen, “Rickettsial actin-based motility: behavior and involvement of cytoskeletal regulators,” Ann. N.Y. Acad. Sci. 990(June 2003):535-547. 312. P.L. McNeil, S.S. Vogel, K. Miyake, M. Terasaki, “Patching plasma membrane disruptions with cytoplasmic membrane,” J. Cell Sci. 113(June 2000):1891. 313. M. Terasaki, K. Miyake, P.L. McNeil, “Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events,” J. Cell Biol. 139(6 October 1997):63-74. 314. T. Togo, J.M. Alderton, G.Q. Bi, R.A. Steinhardt, “The mechanism of facilitated cell membrane resealing,” J. Cell Sci. 112(March 1999):719-731. 315. R.P. Jean, C.S. Chen, A.A. Spector, “Finite-element analysis of the adhesion-cytoskeleton-nucleus mechanotransduction pathway during endothelial cell rounding: axisymmetric model,” J. Biomech. Eng. 127(August 2005):594-600. 316. M.J. Dalby, “Topographically induced direct cell mechanotransduction,” Med. Eng. Phys. 27(November 2005):730-742. 317. J.H. Haga, Y.S. Li, S. Chien, “Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells,” J. Biomech. 40(2007):947-960. 318. Y. Iwata, S. Naito, A. Itai, S. Miyamoto, “Protein structure-based de novo design and synthesis of aldose reductase inhibitors,” Drug Des Discov. 17(2001):349-359. 319. R.I. Christopherson, S.D. Lyons, P.K. Wilson, “Inhibitors of de novo nucleotide biosynthesis as drugs,” Acc. Chem. Res. 35(November 2002):961-971. 320. A. Bhakoo, J.G. Raynes, J.R. Heal, M. Keller, A.D. Miller, “De-novo design of complementary (antisense) peptide mini-receptor inhibitor of interleukin 18 (IL-18),” Mol. Immunol. 41(November 2004):1217-1224. 321. W.J. Cooper, M.L. Waters, “Molecular recognition with designed peptides and proteins,” Curr. Opin. Chem. Biol. 9(December 2005):627-631. 322. K.J. Woycechowsky, K. Vamvaca, D. Hilvert, “Novel enzymes through design and evolution,” Adv. Enzymol. Relat. Areas Mol. Biol. 75(2007):241-294. 323. G.L. Butterfoss, B. Kuhlman, “Computer-based design of novel protein structures,” Annu. Rev. Biophys. Biomol. Struct. 35(2006):49-65. 324. T.L. Haut Donahue, D.C. Genetos, C.R. Jacobs, H.J. Donahue, C.E. Yellowley, “Annexin V disruption impairs mechanically induced calcium signaling in osteoblastic cells,” Bone 35(September 2004):656-663. 325. K.J. Elliot, S.J. Millward-Sadler, M.O. Wright, J.E. Robb, W.H. Wallace, D.M. Salter, “Effects of methotrexate on human bone cell responses to mechanical stimulation,” Rheumatology (Oxford) 43(October 2004):1226-1231. 326. A. Kumar, N. Khandelwal, R. Malya, M.B. Reid, A.M. Boriek, “Loss of dystrophin causes aberrant mechanotransduction in skeletal muscle fibers,” FASEB J. 18(January 2004):102-113. 327. Y. Liu, B.P. Chen, M. Lu, Y. Zhu, M.B. Stemerman, S. Chien, J.Y. Shyy, “Shear stress activation of SREBP1 in endothelial cells is mediated by integrins,” Arterioscler. Thromb. Vasc. Biol. 22(January 2002):76-81. 328. L.D. Alexander, S. Alagarsamy, J.G. Douglas, “Cyclic stretch-induced cPLA2 mediates ERK 1/2 signaling in rabbit proximal tubule cells,” Kidney Int. 65(February 2004):551-563. 329. M. Chiquet, A.S. Renedo, F. Huber, M. Fluck, “How do fibroblasts translate mechanical signals into changes in extracellular matrix production?” Matrix Biol. 22(March 2003):73-80. 330. K.P. Grembowicz, D. Sprague, P.L. McNeil, “Temporary disruption of the plasma membrane is required for c-fos expression in response to mechanical stress,” Mol. Biol. Cell. 10(April 1999):1247-1257. 331. L.H. Fang, Y.H. Zhang, J.J. Ma, G.H. Du, B.S. Ku, H.Y. Yao, Y.P. Yun, T.J. Kim, “Inhibitory effects of tetrandrine on the serum- and platelet-derived growth factor-BB-induced proliferation of rat aortic smooth muscle cells through inhibition of cell cycle progression, DNA synthesis, ERK1/2 activation and c-fos expression,” Atherosclerosis 174(June 2004):215-223. 332. Y.H. Zhang, L.H. Fang, B.S. Ku, “Fangchinoline inhibits rat aortic vascular smooth muscle cell proliferation and cell cycle progression through inhibition of ERK1/2 activation and c-fos expression,” Biochem. Pharmacol. 66(1 November 2003):1853-1860. 333. S. Lazar, E. Gershon, N. Dekel, “Selective degradation of cyclin B1 mRNA in rat oocytes by RNA interference (RNAi),” J. Mol. Endocrinol. 33(August 2004):73-85. 334. M.B. Elowitz, M.G. Surette, P.E. Wolf, J.B. Stock, S. Leibler, “Protein mobility in the cytoplasm of Escherichia coli,” J. Bacteriol. 181(January 1999):197-203. 335. M.D. McGee, R. Rillo, A.S. Anderson, D.A. Starr, “UNC-83 IS a KASH protein required for nuclear migration and is recruited to the outer nuclear membrane by a physical interaction with the SUN protein UNC-84,” Mol. Biol. Cell. 17(April 2006):1790-1801. 336. Q. Zhang, J.N. Skepper, F. Yang, J.D. Davies, L. Hegyi, R.G. Roberts, P.L. Weissberg, J.A. Ellis, C.M. Shanahan, “Nesprins: a novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues,” J. Cell Sci. 114(December 2001):4485-4498. 337. M. Crisp, Q. Liu, K. Roux, J.B. Rattner, C. Shanahan, B. Burke, P.D. Stahl, D. Hodzic, “Coupling of the nucleus and cytoplasm: role of the LINC complex,” J. Cell Biol. 172(2 January 2006):41-53. 337a. P.M. Ferree, H.M. Frydman, J.M. Li, J. Cao, E. Wieschaus, W. Sullivan, “Wolbachia utilizes host microtubules and dynein for anterior localization in the Drosophila oocyte,” PLoS Pathog. 1(14 October 2005):e14. 338. C. Vedrenne, H.P. Hauri, “Morphogenesis of the endoplasmic reticulum: beyond active membrane expansion,” Traffic 7(June 2006):639-646. 339. M. Rakowska, R. Jasinska, J. Lenart, I. Komanska, P. Makowski, A. Dygas, S. Pikula, “Membrane integrity and phospholipid movement influence the base exchange reaction in rat liver microsomes,” Mol. Cell. Biochem. 168(March 1997):163-176. 340. N. Benlimame, D. Simard, I.R. Nabi, “Autocrine motility factor receptor is a marker for a distinct membranous tubular organelle,” J. Cell Biol. 129(April 1995):459-471. 341. B. Burke, J. Ellenberg, “Remodelling the walls of the nucleus,” Nature Rev. Mol. Cell Biol. 3(July 2002):487-497. 342. I. Kiseleva, A. Kamkin, P. Kohl, M.J. Lab, “Calcium and mechanically induced potentials in fibroblasts of rat atrium,” Cardiovasc. Res. 32(July 1996):98-111. 343. M. Nagasawa, M. Kanzaki, Y. Iino, Y. Morishita, I. Kojima, “Identification of a novel chloride channel expressed in the endoplasmic reticulum, Golgi apparatus, and nucleus,” J. Biol. Chem. 276(8 June 2001):20413-20418. 344. G.L. Koch, “Reticuloplasmins: a novel group of proteins in the endoplasmic reticulum,” J. Cell Sci. 87(May 1987):491-492. 345. B. Storrie, W. Yang, “Dynamics of the interphase mammalian Golgi complex as revealed through drugs producing reversible Golgi disassembly,” Biochim. Biophys. Acta 1404(14 August 1998):127-137. 346. W. Muranyi, J. Haas, M. Wagner, G. Krohne, U.H. Koszinowski, “Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina,” Science 297(2 August 2002):854-857. 347. C.M. de Noronha, M.P. Sherman, H.W. Lin, M.V. Cavrois, R.D. Moir, R.D. Goldman, W.C. Greene, “Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr,” Science 294(2 November 2001):1105-1108. 348. T.T. Puck, A. Krystosek, “Role of the cytoskeleton in genome regulation and cancer,” Intl. Rev. Cytology 132(1992):75-108. 349. A.J. Maniotis, C.S. Chen, D.E. Ingber, “Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure,” Proc. Natl. Acad. Sci. (USA) 94(4 February 1997):849-854. 350. L.T. Holth, D.N. Chadee, V.A. Spencer, S.K. Samuel, J.R. Safneck, J.R. Davie, “Chromatin, nuclear matrix and the cytoskeleton: role of cell structure in neoplastic transformation,” Int. J. Oncol. 13(October 1998):827-837. 351. K. Djabali, “Cytoskeletal proteins connecting intermediate filaments to cytoplasmic and nuclear periphery,” Histol. Histopathol. 14(April 1999):501-509. 352. H.S. Barbosa, M.F. Ferreira-Silva, E.V. Guimaraes, L. Carvalho, R.M. Rodrigues, “Absence of vacuolar membrane involving Toxoplasma gondii during its intranuclear localization,” J. Parasitol. 91(February 2005):182-184. 353. H. El Alaoui, S.J. Gresoviac, C.P. Vivares, “Occurrence of the microsporidian parasite Nucleospora salmonis in four species of salmonids from the Massif Central of France,” Folia Parasitol (Praha) 53(March 2006):37-43. 354. Z. Pecka, “Intranuclear development of asexual and sexual generations of Eimeria hermani Farr, 1953, the coccidian parasite of geese,” Zentralbl. Bakteriol. 278(June 1993):570-576. 355. S. Cohen, A.R. Behzad, J.B. Carroll, N. Pante, “Parvoviral nuclear import: bypassing the host nuclear-transport machinery,” J. Gen. Virol. 87(November 2006):3209-3213. 356. V.P. Skulachev, L.E. Bakeeva, B.V. Chernyak, L.V. Domnina, A.A. Minin, O.Y. Pletjushkina, V.B. Saprunova, I.V. Skulachev, V.G. Tsyplenkova, J.M. Vasiliev, L.S. Yaguzhinsky, D.B. Zorov, “Thread-grain transition of mitochondrial reticulum as a step of mitoptosis and apoptosis,” Mol. Cell. Biochem. 256-257(January-February 2004):341-358. 357. K.N. Dahl, A.J. Engler, J.D. Pajerowski, D.E. Discher, “Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures,” Biophys. J. 89(October 2005):2855-2864. 358. K.N. Dahl, S.M. Kahn, K.L. Wilson, D.E. Discher, “The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber,” J. Cell Sci. 117(15 September 2004):4779-4786. 359. W. Cheng, J. Kajstura, J.A. Nitahara, B. Li, K. Reiss, Y. Liu, W.A. Clark, S. Krajewski, J.C. Reed, G. Olivetti, P. Anversa, “Programmed myocyte cell death affects the viable myocardium after infarction in rats,” Exp. Cell Res. 226(1 August 1996):316-327. 360. G. Olivetti, F. Quaini, R. Sala, C. Lagrasta, D. Corradi, E. Bonacina, S.R. Gambert, E. Cigola, P. Anversa, “Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart,” J. Mol. Cell. Cardiol. 28(September 1996):2005-2016. 361. W.P. Roos, B. Kaina, “DNA damage-induced cell death by apoptosis,” Trends Mol. Med. 12(September 2006):440-450. 362. M. Dundr, T. Misteli, “Functional architecture in the cell nucleus,” Biochem. J. 356(1 June 2001):297-310; http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=1221839&blobtype=pdf 363. M. Ohara, T. Hayashi, Y. Kusunoki, M. Miyauchi, T. Takata, M. Sugai, “Caspase-2 and caspase-7 are involved in cytolethal distending toxin-induced apoptosis in Jurkat and MOLT-4 T-cell lines,” Infect. Immun. 72(February 2004):871-879; http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=14742531 364. N. Shah, R.J. Asch, A.S. Lysholm, T.W. Lebien, “Enhancement of stress-induced apoptosis in B-lineage cells by caspase-9 inhibitor,” Blood 104(1 November 2004):2873-2878; http://www.bloodjournal.org/cgi/content/full/104/9/2873 365. Peptide Research Institute, Inc., “List of Substrates and Inhibitors for Apoptotic Cell Death Research,” 2007; http://www.peptide.co.jp/eng/i/index.html 366. M.L. Bajt, J.A. Lawson, S.L. Vonderfecht, J.S. Gujral, H. Jaeschke, “Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a postmitochondrial processing of caspase-8,” Toxicol. Sci. 58(November 2000):109-117. 367. A. Skaletskaya, L.M. Bartle, T. Chittenden, A.L. McCormick, E.S. Mocarski, V.S. Goldmacher, “A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation,” Proc. Natl. Acad. Sci. (USA) 98(3 July 2001):7829-7834. 368. D. Himeji, T. Horiuchi, H. Tsukamoto, K. Hayashi, T. Watanabe, M. Harada, “Characterization of caspase-8L: a novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade,” Blood 99(1 June 2002):4070-4078. 369. C. Skurk, H. Maatz, H.S. Kim, J. Yang, M.R. Abid, W.C. Aird, K. Walsh, “The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP,” J. Biol. Chem. 279(9 January 2004):1513-1525; http://www.jbc.org/cgi/content/full/279/2/1513 370. B. Wagenknecht, M. Hermisson, P. Groscurth, P. Liston, P.H. Krammer, M. Weller, “Proteasome inhibitor-induced apoptosis of glioma cells involves the processing of multiple caspases and cytochrome c release,” J. Neurochem. 75(December 2000):2288-2297. 371. D. Vucic, M.C. Franklin, H.J. Wallweber, K. Das, B.P. Eckelman, H. Shin, L.O. Elliott, S. Kadkhodayan, K. Deshayes, G.S. Salvesen, W.J. Fairbrother, “Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP,” Biochem. J. 385(1 January 2005):11-20. 372. N. Ozoren, K. Kim, T.F. Burns, D.T. Dicker, A.D. Moscioni, W.S. El-Deiry, “The caspase 9 inhibitor Z-LEHD-FMK protects human liver cells while permitting death of cancer cells exposed to tumor necrosis factor-related apoptosis-inducing ligand,” Cancer Res. 60(15 November 2000):6259-6265.
373. Q. Huang, Q.L. Deveraux, S. Maeda, G.S. Salvesen, H.R. Stennicke, B.D. Hammock, J.C. Reed, “Evolutionary conservation of apoptosis mechanisms: lepidopteran and baculoviral inhibitor of apoptosis proteins are inhibitors of mammalian caspase-9,” Proc. Natl. Acad. Sci. (USA) 97(15 February 2000):1427-1432. 374. R. Filomenko, L. Prevotat, C. Rebe, M. Cortier, J.F. Jeannin, E. Solary, A. Bettaieb, “Caspase-10 involvement in cytotoxic drug-induced apoptosis of tumor cells,” Oncogene 25(7 December 2006):7635-7645. 375. D. Milhas, O. Cuvillier, N. Therville, P. Clave, M. Thomsen, T. Levade, H. Benoist, B. Segui, “Caspase-10 triggers Bid cleavage and caspase cascade activation in FasL-induced apoptosis,” J. Biol. Chem. 280(20 May 2005):19836-19842. 376. W. Lee, D.H. Kim, J.H. Boo, Y.H. Kim, I.S. Park, I. Mook-Jung, “ER stress-induced caspase-12 activation is inhibited by PKC in neuronal cells,” Apoptosis 10(March 2005):407-415. 377. M. Vilatoba, C. Eckstein, G. Bilbao, C.A. Smyth, S. Jenkins, J.A. Thompson, D.E. Eckhoff, J.L. Contreras, “Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis,” Surgery 138(August 2005):342-351. 378. H. Wootz, I. Hansson, L. Korhonen, D. Lindholm, “XIAP decreases caspase-12 cleavage and calpain activity in spinal cord of ALS transgenic mice,” Exp. Cell Res. 312(10 June 2006):1890-1898. 379. B.H. Han, D. Xu, J. Choi, Y. Han, S. Xanthoudakis, S. Roy, J. Tam, J. Vaillancourt, J. Colucci, R. Siman, A. Giroux, G.S. Robertson, R. Zamboni, D.W. Nicholson, D.M. Holtzman, “Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic-ischemic brain injury,” J. Biol. Chem. 277(16 August 2002):30128-30136. 380. Y. Chen, R.J. Kelm Jr., R.C. Budd, B.E. Sobel, D.J. Schneider, “Inhibition of apoptosis and caspase-3 in vascular smooth muscle cells by plasminogen activator inhibitor type-1,” J. Cell. Biochem. 92(1 May 2004):178-188.
381. W. Sun, H. Kimura, N. Hattori, S. Tanaka, S. Matsuyama, K. Shiota, “Proliferation related acidic leucine-rich protein PAL31 functions as a caspase-3 inhibitor,” Biochem. Biophys. Res. Commun. 342(14 April 2006):817-823. 382. O. Nyormoi, Z. Wang, M. Bar-Eli, “Sequence-based discovery of a synthetic peptide inhibitor of caspase 6,” Apoptosis 8(August 2003):371-376. 383. S. Shin, B.J. Sung, Y.S. Cho, H.J. Kim, N.C. Ha, J.I. Hwang, C.W. Chung, Y.K. Jung, B.H. Oh, “An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and –7,” Biochemistry 40(30 January 2001):1117-1123. 384. Y. Suzuki, Y. Nakabayashi, K. Nakata, J.C. Reed, R. Takahashi, “X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes,” J. Biol. Chem. 276(20 July 2001):27058-27063. 385. R.K. Reddy, C. Mao, P. Baumeister, R.C. Austin, R.J. Kaufman, A.S. Lee, “Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation,” J. Biol. Chem. 278(6 June 2003):20915-20924. 386. K. Kivinen, M. Kallajoki, P. Taimen, “Caspase-3 is required in the apoptotic disintegration of the nuclear matrix,” Exp. Cell Res. 311(15 November 2005):62-73. 387. V. Cowling, J. Downward, “Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain,” Cell Death Differ. 9(October 2002):1046-1056. 388. T.E. Allsopp, J. McLuckie, L.E. Kerr, M. Macleod, J. Sharkey, J.S. Kelly, “Caspase 6 activity initiates caspase 3 activation in cerebellar granule cell apoptosis,” Cell Death Differ. 7(October 2000):984-993. 389. A. Kawahara, M. Enari, R.V. Talanian, W.W. Wong, S. Nagata, “Fas-induced DNA fragmentation and proteolysis of nuclear proteins,” Genes Cells 3(May 1998):297-306. 390. O. Schickling, A.H. Stegh, J. Byrd, M.E. Peter, “Nuclear localization of DEDD leads to caspase-6 activation through its death effector domain and inhibition of RNA polymerase I dependent transcription,” Cell Death Differ. 8(December 2001):1157-1168. 391. C.T. Workman, H.C. Mak, S. McCuine, J.B. Tagne, M. Agarwal, O. Ozier, T.J. Begley, L.D. Samson, T. Ideker, “A systems approach to mapping DNA damage response pathways,” Science 312(19 May 2006):1054-1059. 392. A.V. Gudkov, E.A. Komarova, “Prospective therapeutic applications of p53 inhibitors,” Biochem. Biophys. Res. Commun. 331(10 June 2005):726-736. 393. W. Duan, X. Zhu, B. Ladenheim, Q.S. Yu, Z. Guo, J. Oyler, R.G. Cutler, J.L. Cadet, N.H. Greig, M.P. Mattson, “p53 inhibitors preserve dopamine neurons and motor function in experimental Parkinsonism,” Ann. Neurol. 52(November 2002):597-606. 394. N. Pietrancosta, A. Moumen, R. Dono, P. Lingor, V. Planchamp, F. Lamballe, M. Bahr, K.L. Kraus, F. Maina, “Imino-tetrahydro-benzothiazole derivatives as p53 inhibitors: discovery of a highly potent in vivo inhibitor and its action mechanism,” J. Med. Chem. 49(15 June 2006):3645-3652. 395. J. Karlseder, K. Hoke, O.K. Mirzoeva, C. Bakkenist, M.B. Kastan, J.H. Petrini, T. de Lange, “The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response,” PLoS Biol. 2(17 August 2004):E240. 396. A. Lau, K.M. Swinbank, P.S. Ahmed, D.L. Taylor, S.P. Jackson, G.C. Smith, M.J. O’Connor, “Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase,” Nat. Cell Biol. 7(May 2005):493-500. 397. R. Daniel, G. Kao, K. Taganov, J.G. Greger, O. Favorova, G. Merkel, T.J. Yen, R.A. Katz, A.M. Skalka, “Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response,” Proc. Natl. Acad. Sci. (USA) 100(15 April 2003):4778-4783. 398. A.M. Garnerone, D. Cabanes, M. Foussard, P. Boistard, J. Batut, “Inhibition of the FixL sensor kinase by the FixT protein in Sinorhizobium meliloti,” J. Biol. Chem. 274(5 November 1999):32500-32506. 399. J.A. Lesley, C.D. Waldburger, “Repression of Escherichia coli PhoP-PhoQ signaling by acetate reveals a regulatory role for acetyl coenzyme A,” J. Bacteriol. 185(April 2003):2563-2570. 400. A. Morita, J. Zhu, N. Suzuki, A. Enomoto, Y. Matsumoto, M. Tomita, T. Suzuki, K. Ohtomo, Y. Hosoi, “Sodium orthovanadate suppresses DNA damage-induced caspase activation and apoptosis by inactivating p53,” Cell. Death Differ. 13(March 2006):499-511. 401. R.R. Leker, M. Aharonowiz, N.H. Greig, H. Ovadia, “The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha,” Exp. Neurol. 187(June 2004):478-486. 402. P. Scaffidi, T. Misteli, M.E. Bianchi, “Release of chromatin protein HMGB1 by necrotic cells triggers inflammation,” Nature 418(11 July 2002):191-195. 403. J.H. Youn, J.S. Shin, “Nucleocytoplasmic shuttling of hmgb1 is regulated by phosphorylation that redirects it toward secretion,” J. Immunol. 177(1 December 2006):7889-7897. 404. P.J. Verschure, I. Van Der Kraan, J.M. Enserink, M.J. Mone, E.M. Manders, R. Van Driel, “Large-scale chromatin organization and the localization of proteins involved in gene expression in human cells,” J. Histochem. Cytochem. 50(October 2002):1303-1312. 405. P.J. Dimario, “Cell and molecular biology of nucleolar assembly and disassembly,” Int. Rev. Cytol. 239(2004):99-178. 406. J. Heix, A. Vente, R. Voit, A. Budde, T.M. Michaelidis, I. Grummt, “Mitotic silencing of human rRNA synthesis : inactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation,” EMBO J. 17(1998):7373-7381. 407. A. Kuhn, A. Vente, M. Doree, I. Grummt, “Mitotic phosphorylation of the TBP-containing factor SL1 represses ribosomal gene transcription,” J. Mol. Biol. 284(1998):1-5. 408. A.I. Lamond, D.L. Spector, “Nuclear speckles: a model for nuclear organelles,” Nat. Rev. Mol. Cell Biol. 4(August 2003):605-612. 409. J.F. Gui, W.S. Lane, X.-D. Fu, “A serine kinase regulates intracellular localization of splicing factors in the cell cycle,” Nature 369(1994):678-682. 410. T. Haaf, D.C. Ward, “Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure, and dispersion of chromosomal domains,” Exp. Cell Res. 224(10 April 1996):163-173. 411. M. Cioce, A.I. Lamond, “Cajal bodies: a long history of discovery,” Annu. Rev. Cell Dev. Biol. 21(2005):105-131. 412. J.E. Sleeman, L. Trinkle-Mulcahy, A.R. Prescott, S.C. Ogg, A.I. Lamond, “Cajal body proteins SMN and Coilin show differential dynamic behaviour in vivo,” J. Cell Sci. 116(15 May 2003):2039-2050. 413. C. Gribbon, R. Dahm, A.R. Prescott, R.A. Quinlan, “Association of the nuclear matrix component NuMA with the Cajal body and nuclear speckle compartments during transitions in transcriptional activity in lens cell differentiation,” Eur. J. Cell Biol. 81(October 2002):557-566. 414. B. Buendia, A. Santa-Maria, J.C. Courvalin, “Caspase-dependent proteolysis of integral and peripheral proteins of nuclear membranes and nuclear pore complex proteins during apoptosis,” J. Cell Sci. 112(June 1999):1743-1753; http://jcs.biologists.org/cgi/reprint/112/11/1743 415. R. Sattar, S.A. Ali, A. Abbasi, “Molecular mechanism of apoptosis: prediction of three-dimensional structure of caspase-6 and its interactions by homology modeling,” Biochem. Biophys. Res. Commun. 308(29 August 2003):497-504. 416. A.E. Kalinin, M. Aho, J. Uitto, S. Aho, “Breaking the connection: caspase 6 disconnects intermediate filament-binding domain of periplakin from its actin-binding N-terminal region,” J. Invest. Dermatol. 124(January 2005):46-55. 417. J.L. Broers, F.C. Ramaekers, G. Bonne, R.B. Yaou, C.J. Hutchison, “Nuclear lamins: laminopathies and their role in premature ageing,” Physiol. Rev. 86(July 2006):967-1008. 418. S. Galande, L.A. Dickinson, I.S. Mian, M. Sikorska, T. Kohwi-Shigematsu, “SATB1 cleavage by caspase 6 disrupts PDZ domain-mediated dimerization, causing detachment from chromatin early in T-cell apoptosis,” Mol. Cell Biol. 21(August 2001):5591-5604. 419. A.G. Porter, R.U. Janicke, “Emerging roles of caspase-3 in apoptosis,” Cell Death Differ. 6(February 1999):99-104. 420. M. Kipp, B.L. Schwab, M. Przybylski, P. Nicotera, F.O. Fackelmayer, “Apoptotic cleavage of scaffold attachment factor A (SAF-A) by caspase-3 occurs at a noncanonical cleavage site,” J. Biol. Chem. 275(18 February 2000):5031-5036. 421. P. Taimen, M. Kallajoki, “NuMA and nuclear lamins behave differently in Fas-mediated apoptosis,” J. Cell Sci. 116(1 February 2003):571-583. 422. C. Gueth-Hallonet, K. Weber, M. Osborn, “Cleavage of the nuclear matrix protein NuMA during apoptosis,” Exp. Cell Res. 233(25 May 1997):21-24. 423. M. Patre, A. Tabbert, D. Hermann, H. Walczak, H.R. Rackwitz, V.C. Cordes, E. Ferrando-May, “Caspases target only two architectural components within the core structure of the nuclear pore complex,” J. Biol. Chem. 281(13 January 2006):1296-1304. 424. P. Widlak, W.T. Garrard, “Discovery, regulation, and action of the major apoptotic nucleases DFF40/CAD and endonuclease G,” J. Cell Biochem 94(15 April 2005):1078-1087. 425. A. Yoshida, Y. Pommier, T. Ueda, “Endonuclease activation and chromosomal DNA fragmentation during apoptosis in leukemia cells,” Int. J. Hematol. 84(July 2006):31-37. 426. K. Zhu, H. Lu, W. Ying, “Post-treatment with the Ca(2+)-Mg(2+)-endonuclease inhibitor aurintricarboxylic acid prevents peroxynitrite-induced DNA damage and death of murine astrocytes,” Biochem. Biophys. Res. Commun. 344(9 June 2006):881-886. 427. M. Haimsohn, R. Beery, A. Karasik, H. Kanety, A. Geier, “Aurintricarboxylic acid induces a distinct activation of the IGF-I receptor signaling within MDA-231 cells,” Endocrinology 143(March 2002):837-845. 428. S. Miscia, A. Di Baldassarre, R. Alba Rana, R. Di Pietro, A. Cataldi, “Engagement of DNA polymerases during apoptosis,” Cell Prolif. 30(August-September 1997):325-340. 429. G. Natale, D. Cattano, A. Abramo, F. Forfori, F. Fulceri, F. Fornai, A. Paparelli, F. Giunta, “Morphological evidence that xenon neuroprotects against N-methyl-DL-aspartic acid-induced damage in the rat arcuate nucleus: a time-dependent study,” Ann. N.Y. Acad. Sci. 1074(August 2006):650-658. 430. F. Aumann, F. Lankas, M. Caudron, J. Langowski, “Monte Carlo simulation of chromatin stretching,” Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 73(April 2006):041927. 431. J. Widom, R.L. Baldwin, “Tests of spool models for DNA packaging in phage lambda,” J. Mol. Biol. 171(25 December 1983):419-437.
432. J. Arsuaga, R.K. Tan, M. Vazquez, D.W. Sumners, S.C. Harvey, “Investigation of viral DNA packaging using molecular mechanics models.” Biophys. Chem. 101-102(10 December 2002):475-484.
433. D. Lechardeur, K.J. Sohn, M. Haardt, P.B. Joshi, M. Monck, R.W. Graham, B. Beatty, J. Squire, H. O’Brodovich, G.L. Lukacs, “Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer,” Gene Ther. 6(April 1999):482-497. 434. H. Pollard, G. Toumaniantz, J.L. Amos, H. Avet-Loiseau, G. Guihard, J.P. Behr, D. Escande, “Ca2+-sensitive cytosolic nucleases prevent efficient delivery to the nucleus of injected plasmids,” J. Gene. Med. 3(March-April 2001):153-164. 435. L. Holmgren, A. Szeles, E. Rajnavolgyi, J. Folkman, G. Klein, I. Ernberg, K.I. Falk, “Horizontal transfer of DNA by the uptake of apoptotic bodies,” Blood 93(1 June 1999):3956-3963.
436. A. Bergsmedh, A. Szeles, M. Henriksson, A. Bratt, M.J. Folkman, A.L. Spetz, L. Holmgren, “Horizontal transfer of oncogenes by uptake of apoptotic bodies,” Proc. Natl. Acad. Sci. (USA) 98(22 May 2001):6407-6411. 437. A. Bergsmedh, A. Szeles, A.L. Spetz, L. Holmgren, “Loss of the p21(Cip1/Waf1) cyclin kinase inhibitor results in propagation of horizontally transferred DNA,” Cancer Res. 62(15 January 2002):575-579. 438. L.J. Wong, D.J. Sharpe, “Regulation of nuclear histone acetyltransferase by nucleic acids, histone-DNA complex, and chromatin,” Biochem. Genet. 29(February 1991):13-28. 439. M. Poirier, S. Eroglu, D. Chatenay, J.F. Marko, “Reversible and irreversible unfolding of mitotic newt chromosomes by applied force,” Mol. Biol. Cell 11(January 2000):269-276. 440. T. Cremer, G. Kreth, H. Koester, R.H. Fink, R. Heintzmann, M. Cremer, I. Solovei, D. Zink, C. Cremer, “Chromosome territories, interchromatin domain compartment, and nuclear matrix: an integrated view of the functional nuclear architecture,” Crit. Rev. Eukaryot. Gene Expr. 10(2000):179-212. 441. A. Bolzer, G. Kreth, I. Solovei, D. Koehler, K. Saracoglu, C. Fauth, S. Muller, R. Eils, C. Cremer, M.R. Speicher, T. Cremer, “Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes,” PLoS Biol. 3(May 2005):e157. 442. G. Kreth, J. Finsterle, J. von Hase, M. Cremer, C. Cremer, “Radial arrangement of chromosome territories in human cell nuclei: a computer model approach based on gene density indicates a probabilistic global positioning code,” Biophys. J. 86(May 2004):2803-2812; http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=15111398 443. R. Mayer, A. Brero, J. von Hase, T. Schroeder, T. Cremer, S. Dietzel, “Common themes and cell type specific variations of higher order chromatin arrangements in the mouse,” BMC Cell Biol. 6(7 December 2005):44. 444. M.R. Branco, A. Pombo, “Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations,” PLoS Biol. 4(May 2006):e138. 445. I. Thomson, S. Gilchrist, W.A. Bickmore, J.R. Chubb, “The radial positioning of chromatin is not inherited through mitosis but is established de novo in early G1,” Curr. Biol. 14(20 January 2004):166-172. 446. A.M. Boutanaev, L.M. Mikhaylova, D.I. Nurminsky, “The pattern of chromosome folding in interphase is outlined by the linear gene density profile,” Mol. Cell Biol. 25(September 2005):8379-8386. 447. N.V. Petrova, O.V. Iarovaia, V.A. Verbovoy, S.V. Razin, “Specific radial positions of centromeres of human chromosomes X, 1, and 19 remain unchanged in chromatin-depleted nuclei of primary human fibroblasts: evidence for the organizing role of the nuclear matrix,” J. Cell. Biochem. 96(1 November 2005):850-857. 448. V. Graziano, S.E. Gerchman, V. Ramakrishnan, “Reconstitution of chromatin higher-order structure from histone H5 and depleted chromatin,” J. Mol. Biol. 203(20 October 1988):997-1007. 449. J.O. Thomas, C. Rees, E.C. Pearson, “Histone H5 promotes the association of condensed chromatin fragments to give pseudo-higher-order structures,” Eur. J. Biochem. 147(15 February 1985):143-151. 450. H. Tanabe, F.A. Habermann, I. Solovei, M. Cremer, T. Cremer, “Non-random radial arrangements of interphase chromosome territories: evolutionary considerations and functional implications,” Mutat. Res. 504(25 July 2002):37-45. 451. M.F. Lyon, “Gene action in the X chromosome of the mouse,” Nature 190(1961):372-373. 452. C. Francastel, D. Schuebler, D.I.K. Martin, M. Groudine, “Nuclear compartmentalization and gene activity,” Nat. Rev. Mol. Cell Biol. 1(2000):137-143. 453. M. Cremer, J. von Hase, T. Volm, A. Brero, G. Kreth, J. Walter, C. Fischer, I. Solovei, C. Cremer, T. Cremer, “Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells,” Chromosome Res. 9(2001):541-567. 454. L.S. Shopland, C.R. Lynch, K.A. Peterson, K. Thornton, N. Kepper, J. Hase, S. Stein, S. Vincent, K.R. Molloy, G. Kreth, C. Cremer, C.J. Bult, T.P. O’Brien, “Folding and organization of a contiguous chromosome region according to the gene distribution pattern in primary genomic sequence,” J. Cell Biol. 174(3 July 2006):27-38. 455. S. Boyle, S. Gilchrist, J.M. Bridger, N.L. Mahy, J.A. Ellis, W.A. Bickmore, “The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells,” Hum. Mol. Genet. 10(1 February 2001):211-219. 456. H. Tanabe, K. Kupper, T. Ishida, M. Neusser, H. Mizusawa, “Inter- and intra-specific gene-density-correlated radial chromosome territory arrangements are conserved in Old World monkeys,” Cytogenet. Genome Res. 108(2005):255-261. 457. L. Mora, I. Sanchez, M. Garcia, M. Ponsa, “Chromosome territory positioning of conserved homologous chromosomes in different primate species,” Chromosoma 115(2006):367-375. 458. M. Ferguson, D.C. Ward, “Cell cycle dependent chromosomal movement in pre-mitotic human T-lymphocyte nuclei,” Chromosoma 101(August 1992):557-565. 459. E.M.M. Manders, A.E. Visser, A. Koppen, W.C. de Leeuw, R. van Liere, G.J. Brakenhoff, R. van Driel, “Four-dimensional imaging of chromatin dynamics during the assembly of the interphase nucleus,” Chromosome Research 11(2003):537-547. 460. K.F. Toth, T.A. Knoch, M. Wachsmuth, M. Frank-Stohr, M. Stohr, C.P. Bacher, G. Muller, K. Rippe, “Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin,” J. Cell Sci. 117(15 August 2004):4277-4287. 461. H. Tamada, N. Van Thuan, P. Reed, D. Nelson, N. Katoku-Kikyo, J. Wudel, T. Wakayama, N. Kikyo, “Chromatin decondensation and nuclear reprogramming by nucleoplasmin,” Mol. Cell Biol. 26(February 2006):1259-1271. 462. G. Vogt, “In vivo decondensation of chromatin and nucleolar fibrillar component by Leucaena leucocephala ingredient,” Biol. Cell. 72(1991):211-215. 463. Z. Karetsou, G. Martic, S. Tavoulari, S. Christoforidis, M. Wilm, C. Gruss, T. Papamarcaki, “Prothymosin alpha associates with the oncoprotein SET and is involved in chromatin decondensation,” FEBS Lett. 577(19 November 2004):496-500. 464. S.Z. Wang, R. Adler, “Chromokinesin: a DNA-binding, kinesin-like nuclear protein,” J. Cell Biol. 128(March 1995):761-768. 465. Y.M. Lee, S. Lee, E. Lee, H. Shin, H. Hahn, W. Choi, W. Kim, “Human kinesin superfamily member 4 is dominantly localized in the nuclear matrix and is associated with chromosomes during mitosis,” Biochem. J. 360(15 December 2001):549-556; http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=1222256&blobtype=pdf 466. A. Zimber, Q.D. Nguyen, C. Gespach, “Nuclear bodies and compartments: functional roles and cellular signalling in health and disease,” Cell Signal 16(October 2004):1085-1104. 467. D.A. Compton, D.W. Cleveland, “NuMA is required for the proper completion of mitosis,” J. Cell Biol. 120(February 1993):947-957. 468. J. Harborth, M. Osborn, “Does NuMA have a scaffold function in the interphase nucleus?” Crit. Rev. Eukaryot. Gene Expr. 9(1999):319-328. 469. O.I. Podgornaya, A.P. Voronin, N.I. Enukashvily, I.V. Matveev, I.B. Lobov, “Structure-specific DNA-binding proteins as the foundation for three-dimensional chromatin organization,” Int. Rev. Cytol. 224(2003):227-296. 470. P. Collas, J.-C. Courvalin, D. Poccia, “Targeting of membranes to sea urchin sperm chromatin is mediated by lamin B receptor-like integral membrane protein,” J. Cell Biol. 135(1996):1715-1725. 471. R. Foisner, L. Gerace, “Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation,” Cell 73(1993):1267-1279. 472. L.J. Thompson, M. Bollen, A.P. Fields, “Identification of protein phosphatase 1 as a mitotic lamin phosphatase,” J. Biol. Chem. 272(21 November 1997):29693-29697. 473. T. Sassa, H. Ueda-Ohba, K. Kitamura, S. Harada, R. Hosono, “Role of Caenorhabditis elegans protein phosphatase type 1, CeGLC-7 beta, in metaphase to anaphase transition during embryonic development,” Exp. Cell Res. 287(15 July 2003):350-360. 474. J.G. Gall, M. Bellini, Z. Wu, C. Murphy, “Assembly of the nuclear transcription and processing machinery: Cajal bodies (coiled bodies) and transcriptosomes,” Mol. Biol. Cell. 10(December 1999):4385-4402. 475. A.I. Lamond, D.L. Spector, “Nuclear speckles: a model for nuclear organelles,” Nat. Rev. Mol. Cell Biol. 4(August 2003):605-612. 476. A.A. Van Hooser, P. Yuh, R. Heald, “The perichromosomal layer,” Chromosoma 114(December 2005):377-388. 477. J.G. Gall, “A role for Cajal bodies in assembly of the nuclear transcription machinery,” FEBS Lett. 498(8 June 2001):164-167. 477a. D. Mijaljica, M. Prescott, R.J. Devenish, “Nibbling within the nucleus: turnover of nuclear contents,” Cell. Mol. Life Sci. 64(March 2007):581-588. 478. M.A. Lever, J.P. Th ‘ng, X. Sun, M.J. Hendzel, “Rapid exchange of histone H1.1 on chromatin in living human cells,” Nature 408(14 December 2000):873-876. 479. N. Fomproix, J. Gebrane-Younes, D. Hernandez-Verdun, “Effects of anti-fibrillarin antibodies on building of functional nucleoli at the end of mitosis,” J. Cell Sci. 111(February 1998):359-372. 480. T. Dousset, C. Wang, C. Verheggen, D. Chen, D. Hernandez-Verdun, S. Huang, “Initiation of nucleolar assembly is independent of RNA polymerase I transcription,” Mol. Biol. Cell. 11(August 2000):2705-2717. 481. G.H. Karpen, J.E. Schaefer, C.D. Laird, “A Drosophila rRNA gene located in euchromatin is active in transcription and nucleolus formation,” Genes Dev. 2(1988):1745-1763. 482. M. Oakes, J.P. Aris, J.S. Brockenbrough, H. Wai, L. Vu, M. Nomura, “Mutational analysis of the structure and localization of the nucleolus in the yeast Saccharomyces cerevisiae,” J. Cell Biol. 143(1998):23-34. 483. S.R. Wente, G. Blobel, “A temperature-sensitive NUP116 null mutant forms a nuclear envelope seal over the yeast nuclear pore complex thereby blocking nucleocytoplasmic traffic,” J. Cell Biol. 123(October 1993):275-284. 484. C. DeHoratius, P.A. Silver. “Nuclear transport defects and nuclear envelope alterations are associated with mutation of the Saccharomyces cerevisiae NPL4 gene,” Mol. Biol. Cell 7(November 1996):1835-1855. 485. C.J. Flickinger, “Interactions between endoplasmic reticulum and nuclear membranes of different types of cells during repair of damaged amoeba nuclei,” Exp. Cell Res. 111(February 1978):427-436. 486. P.R. Clarke, C. Zhang, “Spatial and temporal control of nuclear envelope assembly by Ran GTPase,” Symp. Soc. Exp. Biol. 56(2004):193-204. 487. D.T. Ng, P. Walter, “ER membrane protein complex required for nuclear fusion,” J. Cell Biol. 132(February 1996):499-509. 488. M. Latterich, R. Schekman, “The karyogamy gene KAR2 and novel proteins are required for ER-membrane fusion,” Cell 78(15 July 1994):87-98. 489. T. Barona, R.D. Byrne, T.R. Pettitt, M.J. Wakelam, B. Larijani, D.L. Poccia, “Diacylglycerol induces fusion of nuclear envelope membrane precursor vesicles,” J. Biol. Chem. 280(16 December 2005):41171-41177. 490. K. Kontani, J.H. Rothman, “Cell fusion: EFF is enough,” Curr. Biol. 15(12 April 2005):R252-R254. 491. C.Z. Jin, “Dynamics of membrane skeleton in fused red cell membranes,” J. Cell Sci. 90(May 1988):93-97. 492. H. Takano, S. Kawano, N. Sasaki, H. Kuroiwa, T. Kuroiwa, “Characterization of a putative fusogen encoded in a mitochondrial plasmid of Physarum polycephalum,” J. Plant Res. 115(August 2002):255-261. 493. M. Damek-Poprawa, J. Krouse, C. Gretzula, K. Boesze-Battaglia, “A novel tetraspanin fusion protein, peripherin-2, requires a region upstream of the fusion domain for activity,” J. Biol. Chem. 280(11 March 2005):9217-9224. 494. P.L. McNeil, M. Terasaki, “Coping with the inevitable: how cells repair a torn surface membrane,” Nat. Cell Biol. 3(May 2001):E124-E129. 495. L. Yang, L. Bailey, D. Baltimore, P. Wang, “Targeting lentiviral vectors to specific cell types in vivo,” Proc. Natl. Acad. Sci. (USA) 103(1 August 2006):11479-11484. 496. K. Ramani, Q. Hassan, B. Venkaiah, S.E. Hasnain, D.P. Sarkar, “Site-specific gene delivery in vivo through engineered Sendai viral envelopes,” Proc. Natl. Acad. Sci. (USA) 95(29 September 1998):11886-11890. 497. C.D. Buckley, E.A. Ross, H.M. McGettrick, C.E. Osborne, O. Haworth, C. Schmutz, P.C. Stone, M. Salmon, N.M. Matharu, R.K. Vohra, G.B. Nash, G.E. Rainger, “Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration,” J. Leukoc. Biol. 79(February 2006):303-311. 498. H. Korr, B. Schultze, W. Maurer, “Autoradiographic investigations of glial proliferation in the brain of adult mice. II. Cycle time and mode of proliferation of neuroglia and endothelial cells,” J. Comp. Neurol. 160(15 April 1975):477-490. 499. S.C. Cox, D.M. Walker, “Epithelial growth fraction and expression of p53 tumour suppressor gene in oral submucous fibrosis,” Aust. Dent. J. 41(April 1996):91-96. 500. M. Takatoku, S. Sellers, B.A. Agricola, M.E. Metzger, I. Kato, R.E. Donahue, C.E. Dunbar, “Avoidance of stimulation improves engraftment of cultured and retrovirally transduced hematopoietic cells in primates,” J. Clin. Invest. 108(August 2001):447-455. 501. J. Post, C.Y. Huang, J. Hoffman, “The replication time and pattern of the liver cell in the growing rat,” J. Cell Biol. 18(July 1963):1-12. 502. H. Tsujihashi, A. Nakanishi, H. Matsuda, S. Uejima, T. Kurita, “Growth fraction of human bladder tumors,” Urol. Res. 19(1991):215-218. 503. M. Heenen, P. Galand, “The growth fraction of normal human epidermis,” Dermatology 194(1997):313-317. 504. R. Przemioslo, N.A. Wright, G. Elia, P.J. Ciclitira, “Analysis of crypt cell proliferation in coeliac disease using MI-B1 antibody shows an increase in growth fraction,” Gut 36(January 1995):22-27. 505. T. Heinrich, C. Prowald, R. Friedl, B. Gottwald, R. Kalb, K. Neveling, S. Herterich, H. Hoehn, D. Schindler, “Exclusion/confirmation of Ataxia-telangiectasia via cell-cycle testing,” Eur. J. Pediatr. 165(April 2006):250-257. 506. P. Chatrath, I.S. Scott, L.S. Morris, R.J. Davies, K. Bird, S.L. Vowler, N. Coleman, “Immunohistochemical estimation of cell cycle phase in laryngeal neoplasia,” Br. J. Cancer 95(7 August 2006):314-321. 507. C.E. Bacchi, A.M. Gown, “Detection of cell proliferation in tissue sections,” Braz. J. Med. Biol. Res. 26(July 1993):677-687. 508. K. Oka, T. Nakano, T. Arai, “Cytoplasmic p105 index is an accurate mitotic index, but is not related to prognosis in cervical carcinoma,” Arch. Pathol. Lab. Med. 118(May 1994):506-509. 509. D. Uberti, L. Piccioni, M. Cadei, P. Grigolato, V. Rotter, M. Memo, “p53 is dispensable for apoptosis but controls neurogenesis of mouse dentate gyrus cells following gamma-irradiation,” Brain Res. Mol. Brain Res. 93(10 September 2001):81-89. 510. A. Varis, A.L. Salmela, M.J. Kallio, “Cenp-F (mitosin) is more than a mitotic marker,” Chromosoma 115(August 2006):288-295. 511. C. Giacinti, A. Giordano, “RB and cell cycle progression,” Oncogene 25(28 August 2006):5220-5227. 512. I. Perez-Roger, C. Ivorra, A. Diez, M.J. Cortes, E. Poch, S.M. Sanz-Gonzalez, V. Andres, “Inhibition of cellular proliferation by drug targeting of cyclin-dependent kinases,” Curr. Pharm. Biotechnol. 1(July 2000):107-116. 513. P. Thommes, J.J. Blow, “The DNA replication licensing system,” Cancer Surv. 29(1997):75-90. 514. J.D. Black, “Protein kinase C-mediated regulation of the cell cycle,” Front. Biosci. 5(1 April 2000):D406-D423. 515. G.R. Stark, W.R. Taylor, “Control of the G2/M transition,” Mol. Biotechnol. 32(March 2006):227-248. 516. M. Savio, M. Cerri, O. Cazzalini, P. Perucca, L.A. Stivala, P. Pichierri, A. Franchitto, L. Meijer, E. Prosperi, “Replication-dependent DNA damage response triggered by roscovitine induces an uncoupling of DNA replication proteins,” Cell Cycle 5(September 2006):2153-2159. 517. S. Tada, “Cdt1 and geminin: role during cell cycle progression and DNA damage in higher eukaryotes,” Front. Biosci. 12(1 January 2007):1629-1641. 518. M. Fujita, “Cdt1 revisited: complex and tight regulation during the cell cycle and consequences of deregulation in mammalian cells,” Cell Div. 1(17 October 2006):22. 519. S. Shreeram, A. Sparks, D.P. Lane, J.J. Blow, “Cell type-specific responses of human cells to inhibition of replication licensing,” Oncogene 21(26 September 2002):6624-6632. 520. M.A. Madine, M. Swietlik, C. Pelizon, P. Romanowski, A.D. Mills, R.A. Laskey, “The roles of the MCM, ORC, and Cdc6 proteins in determining the replication competence of chromatin in quiescent cells,” J. Struct. Biol. 129(April 2000):198-210. 521. H. Yoshida, Y. Okada, H. Maruiwa, K. Fukuda, M. Nakamura, K. Chiba, Y. Toyama, “Synaptic blockade plays a major role in the neural disturbance of experimental spinal cord compression,” J. Neurotrauma. 20(December 2003):1365-1376. 522. M. Arundine, G.K. Chopra, A. Wrong, S. Lei, M.M. Aarts, J.F. MacDonald, M. Tymianski, “Enhanced vulnerability to NMDA toxicity in sublethal traumatic neuronal injury in vitro,” J. Neurotrauma. 20(December 2003):1377-1395. 523. A. Campos-Torres, M. Touret, P.P. Vidal, S. Barnum, C. de Waele, “The differential response of astrocytes within the vestibular and cochlear nuclei following unilateral labyrinthectomy or vestibular afferent activity blockade by transtympanic tetrodotoxin injection in the rat,” Neuroscience 130(2005):853-865. 524. P.V. Bayly, K.T. Dikranian, E.E. Black, C. Young, Y.Q. Qin, J. Labruyere, J.W. Olney, “Spatiotemporal evolution of apoptotic neurodegeneration following traumatic injury to the developing rat brain,” Brain Res. 1107(30 August 2006):70-81. 525. D.H. Cormack, Ham’s Histology, J.B. Lippincott Company, Philadelphia PA, 1987. 526. A.O. Dourda, A.J. Moule, W.G. Young, “A morphometric analysis of the cross-sectional area of dentine occupied by dentinal tubules in human third molar teeth,” Int. Endod. J. 27(July 1994):184-189. 527. J. Arends, I. Stokroos, W.G. Jongebloed, J. Ruben, “The diameter of dentinal tubules in human coronal dentine after demineralization and air drying. A combined light microscopy and SEM study,” Caries Res. 29(1995):118-121. 528. J. Black, Biological Performance of Materials: Fundamentals of Biocompatibility, Third Edition, Marcel Dekker, New York, 1999. 529. S.D. Kirley, B.R. Rueda, D.C. Chung, L.R. Zukerberg, “Increased growth rate, delayed senescense and decreased serum dependence characterize cables-deficient cells,” Cancer Biol. Ther. 4(June 2005):654-658. 530. P. Weissman-Shomer, M. Fry, “Chick embryo fibroblasts senscence in vitro: pattern of cell division and life span as a function of cell density,” Mech. Ageing Dev. 4(March-April 1975):159-166. 531. J.R. Hill, Q.A. Winger, R.C. Burghardt, M.E. Westhusin, “Bovine nuclear transfer embryo development using cells derived from a cloned fetus,” Anim. Reprod. Sci. 67(3 July 2001):17-26. 532. S.E. Swanberg, M.E. Delany, “Dynamics of telomere erosion in transformed and non-transformed avian cells in vitro,” Cytogenet. Genome Res. 102(2003):318-325. 533. M. Ogawa, A.C. LaRue, C.J. Drake, “Hematopoietic origin of fibroblasts/myofibroblasts: Its pathophysiologic implications,” Blood 108(1 November 2006):2893-2896. 534. K. Yamaji, H. Tsuda, H. Hashimoto, “Current topics on cytapheresis technologies,” Ther. Apher. 5(August 2001):287-292. 535. J.W. Chan, D.S. Taylor, T. Zwerdling, S.M. Lane, K. Ihara, T. Huser, “Micro-Raman spectroscopy detects individual neoplastic and normal hematopoietic cells,” Biophys. J. 90(15 January 2006):648-656. 536. C.A. Cardasis, G.W. Cooper, “An analysis of nuclear numbers in individual muscle fibers during differentiation and growth: a satellite cell-muscle fiber growth unit,” J. Exp. Zool. 191(March 1975):347-358. 537. J.C. Bruusgaard, K. Liestol, M. Ekmark, K. Kollstad, K. Gundersen, “Number and spatial distribution of nuclei in the muscle fibres of normal mice studied in vivo,” J. Physiol. 551(1 September 2003):467-478. 538. B.J. Payne, L.Z. Saunders, “Heavy metal nephropathy of rodents,” Vet. Pathol. 15(August 1978):51-87 (Suppl 5). 539. E.A. Lock, C.J. Reed, “Trichloroethylene: mechanisms of renal toxicity and renal cancer and relevance to risk assessment,” Toxicol. Sci. 91(June 2006):313-331. 540. P. Liptak, E. Kemeny, B. Ivanyi, “Primer: histopathology of polyomavirus-associated nephropathy in renal allografts,” Nat. Clin. Pract. Nephrol. 2(November 2006):631-636. 541. D. O’Toole, M.F. Raisbeck, “Experimentally induced selenosis of adult mallard ducks: clinical signs, lesions, and toxicology,” Vet. Pathol. 34(July 1997):330-340. 542. P. Pilkington, T. Brown, P. Villegas, B. McMurray, R.K. Page, G.N. Rowland, S.G. Thayer, “Adenovirus-induced inclusion body hepatitis in four-day-old broiler breeders,” Avian Dis. 41(April-June 1997):472-474. 543. D.W. Wilson, M.W. Lame, S.K. Dunston, H.J. Segall, “DNA damage cell checkpoint activities are altered in monocrotaline pyrrole-induced cell cycle arrest in human pulmonary artery endothelial cells,” Toxicol. Appl. Pharmacol. 166(15 July 2000):69-80. 544. M. Spoendlin, H. Moch, F. Brunner, W. Brunner, H.R. Burger, D. Kiss, W. Wegmann, P. Dalquen, M. Oberholzer, G. Thiel, et al., “Karyomegalic interstitial nephritis: further support for a distinct entity and evidence for a genetic defect,” Am. J. Kidney Dis. 25(February 1995):242-252. 545. D. Despommier, W.F. Symmans, R. Dell, “Changes in nurse cell nuclei during synchronous infection with Trichinella spiralis,” J. Parasitol. 77(April 1991):290-295. 546. A.M. Gerdes, M.C. Morales, V. Handa, J.A. Moore, M.R. Alvarez, “Nuclear size and DNA content in rat cardiac myocytes during growth, maturation and aging,” J. Mol. Cell Cardiol. 23(July 1991):833-839. 547. J.S. Chopra, A. Sharma, “Protein energy malnutrition and the nervous system,” J. Neurol. Sci. 110(July 1992):8-20. 548. G.E. Hyde, D. Durham, “Rapid increase in mitochondrial volume in nucleus magnocellularis neurons following cochlea removal,” J. Comp. Neurol. 339(1 January 1994):27-48. 549. D.C. Logan, “The mitochondrial compartment,” J. Exp. Bot. 57(2006):1225-1243. 550. E.S. Lander, H. Lodish, “Mitochondrial diseases: gene mapping and gene therapy,” Cell 61(15 June 1990):925-926. 551. P. Hoeben, “Possible reversal of ageing and other mitochondrial deficiencies through retroviral transfection of mitochondrially encoded proteins to the nucleus,” Med. Hypotheses 41(August 1993):131-133. 552. A.D.N.J. de Grey, The Mitochondrial Free Radical Theory of Aging, R.G. Landes Company, Austin TX, 1999. 553. D.P. Gearing, P. Nagley, “Yeast mitochondrial ATPase subunit 8, normally a mitochondrial gene product, expressed in vitro and imported back into the organelle,” EMBO J. 5(20 December 1986):3651-3655. 554. A.H. Schapira, “Mitochondrial disease,” Lancet 368(1 July 2006):70-82. 555. S.M. Khan, J.P. Bennett Jr., “Development of mitochondrial gene replacement therapy,” J. Bioenerg. Biomembr. 36(August 2004):387-393. 555a. D. Sassera, T. Beninati, C. Bandi, E.A. Bouman, L. Sacchi, M. Fabbi, N. Lo N, “‘Candidatus Midichloria mitochondrii’, an endosymbiont of the tick Ixodes ricinus with a unique intramitochondrial lifestyle,” Int. J. Syst. Evol. Microbiol. 56(November 2006):2535-2540. 556. S. Anderson, A.T. Bankier, B.G. Barrell, M.H. de Bruijn, A.R. Coulson, J. Drouin, I.C. Eperon, D.P. Nierlich, B.A. Roe, F. Sanger, P.H. Schreier, A.J. Smith, R. Staden, I.G. Young, “Sequence and organization of the human mitochondrial genome,” Nature 290(9 April 1981):457-465. 557. L.M. Davids, A.V. Corrigall, P.N. Meissner, “Mitochondrial targeting of human protoporphyrinogen oxidase,” Cell Biol. Int. 30(May 2006):416-426. 558. I. Kissova, M. Deffieu, S. Manon, N. Camougrand, “Uth1p is involved in the autophagic degradation of mitochondria,” J. Biol. Chem. 279(10 September 2004):39068-39074. 559. J.J. Lemasters, “Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging,” Rejuvenation Res. 8(Spring 2005):3-5. 560. M. Gosalvez, J. Diaz-Gil, J. Coloma, L. Salganicoff, “Spectral and metabolic characteristics of mitochondrial fractions from rotenone-induced tumours,” Br. J. Cancer 36(August 1977):243-253. 561. D. Mijaljica, M. Prescott, R.J. Devenish, “Different fates of mitochondria: alternative ways for degradation?” Autophagy 3(January-February 2007):4-9. 562. H.C. Lee, Y.H. Wei, “Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress,” Int. J. Biochem. Cell Biol. 37(April 2005):822-834. 563. M. Patrushev, V. Kasymov, V. Patrusheva, T. Ushakova, V. Gogvadze, A. Gaziev, “Mitochondrial permeability transition triggers the release of mtDNA fragments,” Cell. Mol. Life Sci. 61(December 2004):3100-3103. 564. N. Garcia, J.J. Garcia, F. Correa, E. Chavez, “The permeability transition pore as a pathway for the release of mitochondrial DNA,” Life Sci. 76(29 April 2005):2873-2880. 565. H. Youssoufian, R.E. Pyeritz, “Mechanisms and consequences of somatic mosaicism in humans,” Nat. Rev. Genet. 3(October 2002):748-758. 566. J. Zlotogora, “Germ line mosaicism,” Hum. Genet. 102(April 1998):381-386. 567. C.A. Brenner, H.M. Kubisch, K.E. Pierce, “Role of the mitochondrial genome in assisted reproductive technologies and embryonic stem cell-based therapeutic cloning,” Reprod. Fertil. Dev. 16(2004):743-751. 568. D.C. Adelman, A. Terr, “Allergic and Immunologic Disorders,” in L.M. Tierney, Jr., S.J. McPhee, M.A. Papadakis, eds., Current Medical Diagnosis and Treatment, 35th Edition, Appleton and Lange, Stamford, CT, 1996, pp. 694-718. 569. R.M. Cobb, K.J. Oestreich, O.A. Osipovich, E.M. Oltz, “Accessibility control of V(D)J recombination,” Adv. Immunol. 91(2006):45-109. 570. S. Beck, “Immunogenomics: towards a digital immune system,” Novartis Found. Symp. 254(2003):223-230. 571. A.R. Muotri, V.T. Chu, M.C. Marchetto, W. Deng, J.V. Moran, F.H. Gage, “Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition,” Nature 435(16 June 2005):903-910. 572. K.E. Drexler, Engines of Creation: The Coming Era of Nanotechnology, Anchor Press/Doubleday, New York, 1986. 573. H. Varmus, “Retroviruses,” Science 240(10 June 1988):1427-1435. 574. J. Walter, B. Joffe, A. Bolzer, H. Albiez, P.A. Benedetti, S. Muller, M.R. Speicher, T. Cremer, M. Cremer, I. Solovei, “Towards many colors in FISH on 3D-preserved interphase nuclei,” Cytogenet. Genome Res. 114(2006):367-378. 575. U. Woelfle, E. Breit, K. Zafrakas, M. Otte, F. Schubert, V. Muller, J.R. Izbicki, T. Loning, K. Pantel, “Bi-specific immunomagnetic enrichment of micrometastatic tumour cell clusters from bone marrow of cancer patients,” J. Immunol. Methods 300(May 2005):136-145. 576. P. Friedl, P.B. Noble, P.A. Walton, D.W. Laird, P.J. Chauvin, R.J. Tabah, M. Black, K.S. Zanker, “Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro,” Cancer Res. 55(15 October 1995):4557-4560. 577. B. Molnar, A. Ladanyi, L. Tanko, L. Sreter, Z. Tulassay, “Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients,” Clin. Cancer Res. 7(December 2001):4080-4085. 578. H. Schmidt, G. De Angelis, O. Bettendorf, E. Eltze, A. Semjonow, G. Knichwitz, B. Brandt, “Frequent detection and immunophenotyping of prostate-derived cell clusters in the peripheral blood of prostate cancer patients,” Int. J. Biol. Markers 19(April-June 2004):93-99. 579. J.A. Martin, T.D. Brown, A.D. Heiner, J.A. Buckwalter, “Chondrocyte senescence, joint loading and osteoarthritis,” Clin. Orthop. Relat. Res. 427(October 2004):S96-S103. 580. N.A. Hibberts, A.E. Howell, V.A. Randall, “Balding hair follicle dermal papilla cells contain higher levels of androgen receptors than those from non-balding scalp,” J. Endocrinol. 156(January 1998):59-65. 581. U.R. Mikkelsen, A. Fredsted, H. Gissel, T. Clausen, “Excitation-induced Ca2+ influx and muscle damage in the rat: loss of membrane integrity and impaired force recovery,” J. Physiol. 559(15 August 2004):271-285. 582. H. Nielens, J.P. Devogelaer, J. Malghem, “Occurrence of a painful stress fracture of the femoral neck simultaneously with six other asymptomatic localizations in a runner,” J. Sports Med. Phys. Fitness 34(March 1994):79-82. 583. M. O’Brien, “Exercise and osteoporosis,” Ir. J. Med. Sci. 170(January-March 2001):58-62. 584. K. Asahina, F. Kitahara, M. Yamanaka, T. Akiba, “Influences of excessive exercise on the structure and function of rat organs,” Jpn. J. Physiol. 9(15 September 1959):322-326. 585. D.C. McKenzie, “Markers of excessive exercise,” Can. J. Appl. Physiol. 24(February 1999):66-73. 586. S.N. Davidson, “Technology, medicine & health, Part 7. The age of holistic molecular medicine,” Healthc. Forum J. 38(September-October 1995):80-89. 587. J.R. Sklaroff, “Redundancy management technique for Space Shuttle computers,” IBM J. Res. Develop. 20(January 1976):20-28; http://www.research.ibm.com/journal/rd/201/ibmrd2001E.pdf |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||